Nanoscale Imaging of Synapse Morphology in the Mouse Neocortex in Vivo by Two-Photon STED Microscopy Mirelle Jamilla Tamara Ter Veer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nervous Tissue
Nervous Tissue Prof.Prof. ZhouZhou LiLi Dept.Dept. ofof HistologyHistology andand EmbryologyEmbryology Organization:Organization: neuronsneurons (nerve(nerve cells)cells) neuroglialneuroglial cellscells Function:Function: Ⅰ Neurons 1.1. structurestructure ofof neuronneuron somasoma neuriteneurite a.a. dendritedendrite b.b. axonaxon 1.11.1 somasoma (1)(1) nucleusnucleus LocatedLocated inin thethe centercenter ofof soma,soma, largelarge andand palepale--stainingstaining nucleusnucleus ProminentProminent nucleolusnucleolus (2)(2) cytoplasmcytoplasm (perikaryon)(perikaryon) a.a. NisslNissl bodybody b.b. neurofibrilneurofibril NisslNissl’’ss bodiesbodies LM:LM: basophilicbasophilic massmass oror granulesgranules Nissl’s Body (TEM) EMEM:: RERRER,, freefree RbRb FunctionFunction:: producingproducing thethe proteinprotein ofof neuronneuron structurestructure andand enzymeenzyme producingproducing thethe neurotransmitterneurotransmitter NeurofibrilNeurofibril thethe structurestructure LM:LM: EM:EM: NeurofilamentNeurofilament micmicrotubulerotubule FunctionFunction cytoskeleton,cytoskeleton, toto participateparticipate inin substancesubstance transporttransport LipofuscinLipofuscin (3)(3) CellCell membranemembrane excitableexcitable membranemembrane ,, receivingreceiving stimutation,stimutation, fromingfroming andand conductingconducting nervenerve impulesimpules neurite: 1.2 Dendrite dendritic spine spine apparatus Function: 1.3 Axon axon hillock, axon terminal, axolemma Axoplasm: microfilament, microtubules, neurofilament, mitochondria, -

Abstract Book
Table of Contents Tuesday, May 25, 2021 ............................................................................................................................................................... 5 T1. Astrocyte-Specific Expression of the Extracellular Matrix Gene HtrA1 Regulates Susceptibility to Stress in a Sex- Specific Manner ....................................................................................................................................................................... 5 T2. Plexin-B2 Regulates Migratory Plasticity of Glioblastoma Cells in a 3D-Printed Micropattern Device ............................. 5 T3. Pathoanatomical Mapping of Differential MAPT Expression and Splicing in Progressive Supranuclear Palsy ............... 5 T4. Behavioral Variability in Response to Chronic Stress and Morphine in BXD and Parental Mouse Lines......................... 6 T5. Thyroid-Stimulating Hormone Receptor Regulates Anxiety .............................................................................................. 6 T6. Drugs That Inhibit Microglial Inflammation Also Ameliorate Aβ1-42 Induced Toxicity in C. Elegans ............................... 7 T7. Phosphodiesterase 1b is an Upstream Regulator of a Key Gene Network in the Nucleus Accumbens Driving Addiction- Like Behaviors ......................................................................................................................................................................... 7 T8. Reduced Gap Effect in Children With FOXP1 Syndrome and Autism Spectrum -

Absence of S100A4 in the Mouse Lens Induces an Aberrant Retina-Specific Differentiation Program and Cataract
www.nature.com/scientificreports OPEN Absence of S100A4 in the mouse lens induces an aberrant retina‑specifc diferentiation program and cataract Rupalatha Maddala1*, Junyuan Gao2, Richard T. Mathias2, Tylor R. Lewis1, Vadim Y. Arshavsky1,3, Adriana Levine4, Jonathan M. Backer4,5, Anne R. Bresnick4 & Ponugoti V. Rao1,3* S100A4, a member of the S100 family of multifunctional calcium‑binding proteins, participates in several physiological and pathological processes. In this study, we demonstrate that S100A4 expression is robustly induced in diferentiating fber cells of the ocular lens and that S100A4 (−/−) knockout mice develop late‑onset cortical cataracts. Transcriptome profling of lenses from S100A4 (−/−) mice revealed a robust increase in the expression of multiple photoreceptor‑ and Müller glia‑specifc genes, as well as the olfactory sensory neuron‑specifc gene, S100A5. This aberrant transcriptional profle is characterized by corresponding increases in the levels of proteins encoded by the aberrantly upregulated genes. Ingenuity pathway network and curated pathway analyses of diferentially expressed genes in S100A4 (−/−) lenses identifed Crx and Nrl transcription factors as the most signifcant upstream regulators, and revealed that many of the upregulated genes possess promoters containing a high‑density of CpG islands bearing trimethylation marks at histone H3K27 and/or H3K4, respectively. In support of this fnding, we further documented that S100A4 (−/−) knockout lenses have altered levels of trimethylated H3K27 and H3K4. Taken together, -

Electrical Synapses and Their Functional Interactions with Chemical Synapses
REVIEWS Electrical synapses and their functional interactions with chemical synapses Alberto E. Pereda Abstract | Brain function relies on the ability of neurons to communicate with each other. Interneuronal communication primarily takes place at synapses, where information from one neuron is rapidly conveyed to a second neuron. There are two main modalities of synaptic transmission: chemical and electrical. Far from functioning independently and serving unrelated functions, mounting evidence indicates that these two modalities of synaptic transmission closely interact, both during development and in the adult brain. Rather than conceiving synaptic transmission as either chemical or electrical, this article emphasizes the notion that synaptic transmission is both chemical and electrical, and that interactions between these two forms of interneuronal communication might be required for normal brain development and function. Communication between neurons is required for Electrical and chemical synapses are now known to brain function, and the quality of such communica- coexist in most organisms and brain structures, but details tion enables hardwired neural networks to act in a of the properties and distribution of these two modalities of dynamic fashion. Functional interactions between transmission are still emerging. Most research efforts neurons occur at anatomically identifiable cellular have focused on exploring the mechanisms of chemi- regions called synapses. Although the nature of synaptic cal transmission, and considerably less is known transmission has been an area of enormous controversy about those underlying electrical transmission. It was (BOX 1), two main modalities of synaptic transmission — thought that electrical synapses were more abundant namely, chemical and electrical — are now recognized. At in invertebrates and cold-blooded vertebrates than chemical synapses, information is transferred through in mammals. -

Astroglial Networks Scale Synaptic Activity and Plasticity
Astroglial networks scale synaptic activity and plasticity Ulrike Pannascha, Lydia Vargováb,c, Jürgen Reingruberd, Pascal Ezana, David Holcmand, Christian Giaumea, Eva Sykováb,c, and Nathalie Rouacha,1 aCenter for Interdisciplinary Research in Biology, Centre National de la Recherche Scientifique Unité Mixte de Recherche 7241/Institut National de la Santé et de la Recherche Médicale U1050, Collège de France, 75005 Paris, France; bDepartment of Neuroscience and Center for Cell therapy and Tissue Repair, Charles University, Second Faculty of Medicine, 150 06 Prague, Czech Republic; cDepartment of Neuroscience, Institute of Experimental Medicine of the Academy of Sciences of the Czech Republic, 142 20 Prague, Czech Republic; and dInstitut de Biologie de l’Ecole Normale Supérieure, Centre National de la Recherche Scientifique Unité Mixte de Recherche 8197/Institut National de la Santé et de la Recherche Médicale U1024, Department of Computational Biology and Mathematics, Ecole Normale Supérieure, Paris, 75005, France Edited* by Roger A. Nicoll, University of California, San Francisco, CA, and approved April 11, 2011 (received for review November 22, 2010) Astrocytes dynamically interact with neurons to regulate synaptic during synaptic activity. This impairment results in increased transmission. Although the gap junction proteins connexin 30 neuronal excitability, release probability, glutamate spillover, and (Cx30) and connexin 43 (Cx43) mediate the extensive network insertion of postsynaptic AMPARs, unsilencing synapses. Alto- organization of astrocytes, their role in synaptic physiology is un- gether, our findings indicate that gap junction-mediated astroglial known. Here we show, by inactivating Cx30 and Cx43 genes, that networks control synaptic strength by modulating extracellular astroglial networks tone down hippocampal synaptic transmission homeostasis. -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function. -

11 Introduction to the Nervous System and Nervous Tissue
11 Introduction to the Nervous System and Nervous Tissue ou can’t turn on the television or radio, much less go online, without seeing some- 11.1 Overview of the Nervous thing to remind you of the nervous system. From advertisements for medications System 381 Yto treat depression and other psychiatric conditions to stories about celebrities and 11.2 Nervous Tissue 384 their battles with illegal drugs, information about the nervous system is everywhere in 11.3 Electrophysiology our popular culture. And there is good reason for this—the nervous system controls our of Neurons 393 perception and experience of the world. In addition, it directs voluntary movement, and 11.4 Neuronal Synapses 406 is the seat of our consciousness, personality, and learning and memory. Along with the 11.5 Neurotransmitters 413 endocrine system, the nervous system regulates many aspects of homeostasis, including 11.6 Functional Groups respiratory rate, blood pressure, body temperature, the sleep/wake cycle, and blood pH. of Neurons 417 In this chapter we introduce the multitasking nervous system and its basic functions and divisions. We then examine the structure and physiology of the main tissue of the nervous system: nervous tissue. As you read, notice that many of the same principles you discovered in the muscle tissue chapter (see Chapter 10) apply here as well. MODULE 11.1 Overview of the Nervous System Learning Outcomes 1. Describe the major functions of the nervous system. 2. Describe the structures and basic functions of each organ of the central and peripheral nervous systems. 3. Explain the major differences between the two functional divisions of the peripheral nervous system. -
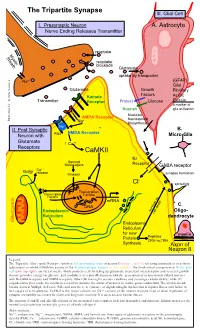
Tripartite Synapse III
The Tripartite Synapse III. Glial Cell I. Presynaptic Neuron A. Astrocyte Nerve Ending Releases Transmitter reuptake Myelin Sheath reuptake blockade Glutamate uptake by transporter Na+ (GFAP) Glial Glutamate Growth Fibrillary Factors Kainate Acidic Transmitter Receptor Protect Glucose Protein a marker of Nourish glia activation Modulate AMPA Receptor Neurosteroid Illustration by: Kendra Scouten Kendra by: Illustration Na+ Biosynthesis Mg++ II. Post Synaptic B. NMDA Receptor Neuron with PSD Micro-Glia Glutamate ↑ Ca++ Receptors CaMKII Bz Second Messengers Receptor GABA receptor ++ Golgi Ca release Kinases synapse formation - Cl sprouting Transcription Transcription Factors Factors mRNA DNA C. Endoplasmic survival vs. Oligo- cell death Reticulum dendrocyte Mitochondria Endoplasmic Reticulum for new myelin Protein Peptides CRH vs.TRH Synthesis Axon of Neuron II Legend: The Tripartite (three-part) Synapse involves: I) a presynaptic axon of neuron I (orange, top left) releasing transmitters to activate (glutamate) or inhibit (GABA-Bz) activity of the II) post-synaptic neuron (yellow, middle). The third critical component is III) the glial cell (pink, top right), an (A) astrocyte which protects cells by taking up glutamate to prevent overexcitation and secretes growth factors; provides energy via glucose; and modulates receptor (R) function with the generation of neurosteroids (which interact with Bz-GABA receptors and NMDA receptors. Other (B) microglia secrete cytokines and scavenge cellular debris; while (C) oligodendrocytes make the myelin necessary to insulate the axons of neurons to insure good conductivity. The myelin sheath breaks down in Multiple Sclerosis (MS) and now there is evidence of oligodendroglia dysfunction in bipolar illness and failure in late stages of schizophrenia. CaMK-II is the major calcium ion (Ca++) sensor of the neuron involved in up or down regulation of synaptic excitability necessary for short and long term memory. -

Neuronal Signaling Neuroscience Fundamentals > the Nerve Cell > the Nerve Cell
Neuronal Signaling Neuroscience Fundamentals > The Nerve Cell > The Nerve Cell NEURONAL SIGNALING SUMMARY ACTION POTENTIALS See: Action Potentials Key Features • All-or-nothing events • Self-propagating • Conduction speed determined by axon diameter (thicker means faster) and amount of myelination (more myelin means faster transmission). CHEMICAL SYNAPSE Steps of Chemical Synapsis 1) The action potential travels down axon of presynaptic neuron. 2) Vesicle fuses with plasma membrane and releases neurotransmitter into the synaptic cleft. 3) Neurotransmitters bind to ligand-gated ion channels, opening them. 4) Ions entering through the open channels cause a depolarization of the membrane, opening voltage-gated ion channels. 5) Ions pass through these voltage-gated ion channels, causing further depolarization of the membrane. 6) This depolarization (action potential) travels along the membrane as more voltage-gated ion channels are opened. ELECTRICAL SYNAPSE • Cytoplasms of adjacent neurons are connected, allowing ions (and action potentials) to travel directly to the next cell. NEURONAL CODING OF STIMULUS STRENGTH Action Potential Frequency • Stimulus strength is based on frequency of action potentials. Action Potential Strength • Strong stimuli can produce another action potential during the relative refractory period. 1 / 7 ABSOLUTE REFRACTORY PERIOD No further action potentials • Voltage-gated sodium channels are open or inactivated so another stimulus, no matter its strength, CANNOT elicit another action potential. RELATIVE REFRACTORY -

Synapse Transmission
Synapse Transmission There are two types of synapses found in your body: electrical and chemical. Electrical synapses allow the direct passage of ions and signaling molecules from cell to cell. In contrast, chemical synapses do not pass the signal directly from the presynaptic cell to the postsynaptic cell. In a chemical synapse, an action potential in the presynaptic neuron leads to the release of a chemical messenger called aneurotransmitter. The neurotransmitter then diffuses across the synapse and binds to receptors on the postsynaptic cell. Binding of the neurotransmitter leads to the production of an electrical signal in the postsynaptic cell. Why does the body have two types of synapses? Each type of synapse has functional advantages and disadvantages. An electrical synapse passes the signal very quickly, which allows groups of cells to act in unison. A chemical synapse takes much longer to transmit the signal from one cell to the next; however, chemical synapses allow neurons to integrate information from multiple presynaptic neurons, determining whether or not the postsynaptic cell will continue to propagate the signal. Neurons respond differently based on information transmitted by multiple chemical synapses. Let’s take a closer look at the structure and function of each type of synapse. Electrical synapses transmit action potentials via the direct flow of electrical current at gap junctions. Gap junctions are formed when two adjacent cells have transmembrane pores that align. The membranes of the two cells are linked together and the aligned pores form a passage between the cells. Consequently, several types of molecules and ions are allowed to pass between the cells. -

Synaptic Transmission Dr
Synaptic Transmission Dr. Simge Aykan Department of Physiology Synaptic Transmission • Biological process by which a neuron communicates with a target cell across a synapse • Synapse is an anatomically specialized junction between two neurons, at which the electrical activity in a presynaptic neuron influences the electrical activity of a postsynaptic neuron • Synapse can be between a neuron and a • Neuron • Muscle • Gland cell Synaptic Transmission • The average neuron forms several thousand synaptic connections and receives a similar number • The Purkinje cell of the cerebellum receives up to 100,000 synaptic inputs • 1011 neurons, 1014 (100 trillion!) synapses Synaptic Transmission • Electrical synapse transmission: transfer of electrical signals through gap junctions • Chemical synaptic transmission: release of a neurotransmitter from the pre-synaptic neuron, and neurotransmitter binding to specific post-synaptic receptors Electrical Synapses • Connection through gap junctions • Narrow gap between membranes (3 nm) • Connexin connexon gap junction • Direct ion passage from one neuron to another • Big enough for many small organic molecules to pass through (1-2 nm) • Mostly between dendrites Electrical Synapses • Electrical postsynaptic potential (PSP) induced by ionic current flow (1 mV or less) Electrical Synapses • Advantages • Extremely rapid • Orchestrating the actions of large groups of neurons • Can transmit metabolic signals between cells • Less common in vertebrate nervous system • Require a large area of contact; restricting -

Tripartite Synapses: Glia, the Unacknowledged Partner
L ETTERS TO THE EDITOR components of the stretch-reflex system that this ‘system’ also includes the major tulated to interconnect and integrate them include: (1) dorsal-root-ganglion cells with sensory and motor systems as well. In a for whatever behavior or function is under their peripheral process that ends in stri- widely used fear-conditioning paradigm, consideration. ated-muscle stretch receptors and their auditory stimuli are used as conditioning Larry Swanson central process that ends on ventral-horn stimuli, foot-shock (somatosensory) stim- Gorica Petrovich motoneurons; and (2) the innervated uli are used as unconditioned stimuli and Neuroscience Program, University of ventral-horn motoneurons themselves. In the behavior of the animal that follows the Southern California, Los Angeles, other words, the stretch-reflex system presentation of such stimuli relies on the CA 90089-2520, USA. itself consists of parts of two classical sys- somatomotor system. In fact, very wide- tems: the somatosensory (proprioceptive) spread parts of the nervous system must References and somatomotor systems. be active during fear conditioning and 1 Lanuza, E., Martínez-Marcos, A. and Martínez-García, F. (1999) Trends Lanuza and colleagues suggest that emotional learning in general. Neurosci. 22, 207 because the basolateral amygdala and cen- Unfortunately, there is no general, 2 Nieuwenhuys, R., ten Donkellar, H.J. tral amygdala are interconnected, and have systematic theory or taxonomy of the and Nicholson, C., eds (1997) The Central Nervous System of Vertebrates, been implicated in fear conditioning and organization of mammalian neural systems. Springer-Verlag emotional learning, these two brain areas The development of one could be a major 3 Swanson, L.W.