Of Low-Density, Fine-Celled Polypropylene Foams
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thieves® Whitening Toothpaste
THIEVES® WHITENING TOOTHPASTE PRODUCT SUMMARY Start your morning on the bright side with the natural power of Thieves® Whitening Toothpaste. Pure and safe ingredients combine to whiten teeth, fight plaque, support healthy gums, and remove stains without damaging enamel. Great for the whole family, Young Living’s exclusive formula is both fluoride free and free of other harsh ingredients, offering you a superior clean you can be confident in. Grin from ear to ear knowing you made a fresh choice with Thieves Whitening Toothpaste! KEY INGREDIENTS Calcium hydroxyapatite, Silica, Perlite, Xylitol, Calcium bicarbonate, Thieves essential oil blend, Spearmint essential oil, Peppermint essential oil, Orange essential oil EXPERIENCE BENEFITS & FEATURES Smile bright with Thieves Whitening Toothpaste. • Whitens and brightens teeth Thieves fans will fall in love with this fresh take on Thieves-infused toothpaste. Fresh Peppermint, • Gently removes surface stains and cleans teeth cool Spearmint, and zesty Orange essential oils • Free from harmful peroxides combine with our spicy Thieves blend to leave your breath EO-fresh all day. Pure and safe whitening • Helps reduce and prevent tartar buildup agents leave you with long-lasting freshness and • Formulated without fluoride, SLS, parabens, a sparkling smile! phthalates, mineral oil, synthetic perfumes or dyes, toxic ingredients, or artificial colors, flavors, or preservatives PRODUCT BACKGROUND • Fights plaque and supports healthy gums Thieves Whitening Toothpaste is one more way Young Living is bringing the power of nature to your • Provides a long-lasting, smooth, fresh feel daily routine: a toothpaste gentle enough for every • Fluoride free and cruelty free day, formulated with essential oils and other natural • Formulated without GMOs ingredients. -

MSDS for Anti-Foaming Agent
MATERIAL SAFETY DATA SHEET for Anti-Foaming Agent Revision: 1.0 1/19/2005 Page 1 of 8 1. PRODUCT AND COMPANY IDENTIFICATION Product Name: Anti-Foaming Agent Chemical Name: Polydimethylsiloxane emulsion Supplier: The Real Milk Paint Co. 11 W. Pumping Station Rd. Quakertown, PA 18951 Contact Numbers: CHEM-TELL (24 Hours) 800-255-0573 Customer Service 800-339-9748 2. COMPOSITION/INFORMATION ON INGREDIENTS COMPOENT CAS3 CONCENTRATION Water 7732-18-5 >50.0% Proprietary additives Trade Secret <30.0% Silica filled polydimethylsiloxane Trade Secret <15.0% Note(s): See Section 15 for chemicals appearing on Federal or State Right-TO-Know lists. 3. HAZARDOUS IDENTIFICATION EMERGENCY OVERVIEW NORMAL PRECAUTIONS COMMON TO SAFE MANUFACTURING PRACTICE SHOULD BE FOLLOWED IN HANDLING AND STORAGE. 4. FIRST AID MEASURES Swallowing No emergency care anticipated Skin Wash skin with soap and water MATERIAL SAFETY DATA SHEET for Anti-Foaming Agent Revision: 1.0 1/19/2005 Page 2 of 8 Inhalation No emergency care anticipated. Eye contact Flush eyes thoroughly with water for several minutes Notes to physicians Toxicology studies have shown this material to be of very low acute toxicity. There is no specific antidote. Treatment of overexposure should be directed at the control of symptoms and the clinical condition of the patient. 5. FIRE FIGHTING MEASURES Flammable limits Lower limits Not available Upper limits Not available Special fire fighting procedures None. Special protective equipment for firefighters Use self-contained breathing apparatus when fighting fires in enclosed areas. Extinguishing media Suitable: Non-flammable (aqueous) After water evaporates, the remaining material will burn. Large fires: -Alcohol-type foam or universal type foam Small fires: -CO2 -dry chemical Unsuitable: None. -

Injection Molding Foam Guidelines
SAFOAM USER’S GUIDE This ‘SAFOAM User’s guide’ was prepared as a service to the customers using Reedy International Corporation’s SAFOAM family of endothermic foaming agents. Reproduction or distribution of this brochure, in whole or in part, is prohibited without the permission of Reedy International Corporation. TABLE OF CONTENTS 1. Introduction ........................................................................................................1 SAFOAM - Its Use in Thermoplastics ........................................................2 2. Structural Plastics and Polymer Selection .........................................................3 Properties of Amorphous Polymers .............................................................3 Properties of Crystalline Polymers ..............................................................4 Properties of Thermoplastic Elastomers ......................................................4 3. Foaming Agent Selection ...................................................................................6 Physical Foaming Agents ............................................................................7 Chemical Foaming Agents ...........................................................................7 Exothermic Chemical Foaming Agents Azodicarbonamide .................................................................................8 Endothermic Chemical Foaming Agents Sodium Borohydride ..............................................................................9 Traditional Acid/Carbonate Systems -
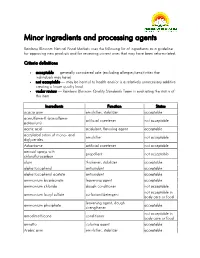
Minor Ingredients and Processing Agents
Minor ingredients and processing agents Rainbow Blossom Natural Food Markets uses the following list of ingredients as a guideline for approving new products and for reviewing current ones that may have been reformulated. Criteria definitions acceptable — generally considered safe (excluding allergies/sensitivities that individuals may have) not acceptable — may be harmul to health and/or is a relatively unnecessary additive creating a lower quality food under review — Rainbow Blossom Quality Standards Team is evaluating the status of this item Ingredients Function Status acacia gum emulsifier, stabilizer acceptable acesulfame-K (acesulfame- artificial sweetener not acceptable potassium) acetic acid acidulant, flavoring agent acceptable acetylated esters of mono- and emulsifier not acceptable diglycerides Advantame artificial sweetener not acceptable aerosol sprays with propellant not acceptable chloroflurocarbon algin thickener, stabilizer acceptable alpha tocopherol antioxidant acceptable alpha tocopherol acetate antioxidant acceptable ammonium bicarbonate leavening agent acceptable ammonium chloride dough conditioner not acceptable not acceptable in ammonium lauryl sulfate surfactant/detergent body care or food leavening agent, dough ammonium phosphate acceptable strengthener not acceptable in amodimethicone conditioner body care or food annatto coloring agent acceptable Arabic gum emulsifier, stabilizer acceptable Ingredients Function Status artificial colors (red #3, red # 40, yellow #5, yellow #6, blue #1, blue coloring agent -

Oleogel-Based Systems for the Delivery of Bioactive Compounds in Foods
gels Review Oleogel-Based Systems for the Delivery of Bioactive Compounds in Foods Tiago C. Pinto 1,2 , Artur J. Martins 2 , Lorenzo Pastrana 2 , Maria C. Pereira 1 and Miguel A. Cerqueira 2,* 1 LEPABE—Laboratory for Process Engineering, Environment, Biotechnology and Energy, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal; [email protected] (T.C.P.); [email protected] (M.C.P.) 2 INL—International Iberian Nanotechnology Laboratory, Avenida Mestre José Veiga, 4715-330 Braga, Portugal; [email protected] (A.J.M.); [email protected] (L.P.) * Correspondence: [email protected] Abstract: Oleogels are semi-solid materials containing a large fraction of liquid oil entrapped in a network of structuring molecules. In the food industry, these formulations can be used to mimic fats and to deliver bioactive compounds. In the last decade, there has been increasing interest in these structures, not only from a scientific point of view, i.e., studying new molecules, methodologies for gelification, and new structures, but also from a technological point of view, with researchers and companies exploring these structures as a way to overcome certain challenges and/or create new and innovative products. One of the exciting applications of oleogels is the delivery of functional molecules, where the incorporation of oil-soluble functional compounds can be explored not only at the macroscale but also at micro- and nanoscales, resulting in different release behaviors and also different applications. This review presents and discusses the most recent works on the development, production, characterization, and applications of oleogels and other oleogel-based systems to deliver functional molecules in foods. -

Industrial Hygiene Characterization of Urea Formaldehyde and Polyurethane Foam Insulation
TECHNICAL REPORT Industrial Hygiene Characterization of Urea Formaldehyde and Polyurethane Foam Insulation U.S. DEPARTMENT OF HEAL TH AND HUMAN SERVICES Public Health Service Centers for Disease Control National Institute for Occupational Safety and Health INDUSTRIAL HYGIENE CHARACTERIZATION OF UREA FORMALDEHYDE AND POLYURETHANE FOAM INSULATION Andrew A. Alcarese Jr. Robert P. Reisdorf Enviro Control, Inc. Rockville, Maryland 20852 Contract No. 210-78-0081 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Centers for Disease Control National Institute for Occupational Safety and Health Division of Surveillance, Hazard Evaluations, and Field Studies Cincinnati, .Ohio 45226 April 1983 DISCLAIMER Ment:Lon of company names or products in this report does not constitute endorsement by the National Institute for Occupational Safety and Health . NIOSH Project Officer: Robert F. Herrick Enviro Control, Inc., Project Manager: Donald W. Rumsey bHHS (NjOSH) Publicatidn No. 83-108 For sale by the Superintendent o ( Doc uments, U .S, Government Printing Office , Waahingtorl, D.C . 20402 .. J. J. ABSTRACT Industrial hygiene surveys were conducted at urea formaldehyde and polyurethane foam thermal insulation manufacturing and application facilities as part of a NIOSH industrywide study of foam insulation materials. In all, eight sites were surveyed--two sets of manufacturers and applicators for each of the two types of foams. Personal and area monitoring were conducted to determine exposure levels to chemical substances and physical agents. Within the urea formaldehyde industries surveyed formaldehyde exposures at the manufacturing facilities ranged from a TWA value of 0.18 ppm to peak values of 5-10 ppm; exposures at the application sites ranged from <0.08 to 2.4 ppm. -

Characterization of Different Chemical Blowing Agents and Their Applicability to Produce Poly(Lactic Acid) Foams by Extrusion
applied sciences Article Characterization of Different Chemical Blowing Agents and Their Applicability to Produce Poly(Lactic Acid) Foams by Extrusion Ákos Kmetty 1,2,* , Katalin Litauszki 1 and Dániel Réti 1 1 Department of Polymer Engineering, Faculty of Mechanical Engineering, Budapest University of Technology and Economics, M˝uegyetemrkp. 3., H-1111 Budapest, Hungary; [email protected] (K.L.); [email protected] (D.R.) 2 MTA–BME Research Group for Composite Science and Technology, M˝uegyetemrkp. 3., H-1111 Budapest, Hungary * Correspondence: [email protected]; Tel.: +36-1-463-2004 Received: 25 September 2018; Accepted: 11 October 2018; Published: 17 October 2018 Abstract: This study presents the applicability of different types (exothermic and endothermic) of chemical blowing agents (CBAs) in the case of poly(lactic acid) (PLA). The amount of foaming agent is a fixed 2 wt%. We used a twin-screw extruder and added the individual components in the form of dry mixture through the hopper of the extruder. We characterized the PLA matrix and the chemical blowing agents with different testing methods. In case of the produced foams we carried out morphological and mechanical tests and used scanning electron microscopy to examine cell structure. We showed that PLA can be successfully foamed with the use of chemical blowing agents. The best results were achieved with an exothermic CBA and with PLA type 8052D. The cell population density of PLA foams produced this way was 4.82 × 105 cells/cm3, their expansion was 2.36, their density 0.53 g/cm3 and their void fraction was 57.61%. -

Antibacterial Efficacy of Novel Eastern Medicine-Inspired Toothpastes
Pacific Journal of Health Volume 3 Issue 1 Article 3 2020 Antibacterial Efficacy of vNo el Eastern Medicine-Inspired Toothpastes Compared to Commercial Formulations Joshua S. Goldfaden University of the Pacific, Arthur A. Dugoni School of Dentistry, [email protected] Follow this and additional works at: https://scholarlycommons.pacific.edu/pjh Part of the Alternative and Complementary Medicine Commons, Dental Hygiene Commons, Dental Materials Commons, Organic Chemicals Commons, and the Pharmaceutical Preparations Commons Recommended Citation Goldfaden, Joshua S. (2020) "Antibacterial Efficacy of vNo el Eastern Medicine-Inspired Toothpastes Compared to Commercial Formulations," Pacific Journal of Health: Vol. 3 : Iss. 1 , Article 3. Available at: https://scholarlycommons.pacific.edu/pjh/vol3/iss1/3 This Article is brought to you for free and open access by Scholarly Commons. It has been accepted for inclusion in Pacific Journal of Health yb an authorized editor of Scholarly Commons. For more information, please contact [email protected]. Antibacterial Efficacy of vNo el Eastern Medicine-Inspired Toothpastes Compared to Commercial Formulations Abstract Toothpaste is an oral health care agent that dates back to the 5th Century B.C. in East Asia. Following the conception of dentistry in the 7th Century B.C. and the field’s subsequent growth, the embodiment of toothpaste has evolved from containing natural substances such as tea leaves to abrasive chemicals, particularly: fluoride, triclosan, and sodium lauryl sulfate (SLS). Regarding how each respective reagent functions, fluoride forms a complex with calcium ions in the teeth ot prevent erosion of the protective enamel coating. This ion also disrupts metabolic activities of Streptococcus mutans (S. -

United States Patent (19) 11 Patent Number: 4,731,193 Mason Et Al
United States Patent (19) 11 Patent Number: 4,731,193 Mason et al. (45) Date of Patent: Mar. 15, 1988 54 AQUEOUSFOAMCONTAINING CHLORINE 56) References Cited DOXDE COMPOSITION AND PREPARATION THEREOF U.S. PATENT DOCUMENTS 2,392,936 1/1946 Mattin et al. ......................... 134/42 2,678,922 5/1954 Stone.......................... 252/187.22 X 75) Inventors: John Y. Mason, Plymouth; Bruce W. 3,278,447 10/1966 McNicholas ............... 252/187.21 X Hicks, Rio Linda; Donald C. English, 4,084,747 4/1978 Alliger ....................... 252/187.23 X Carmichael, all of Calif. 4,104,190 8/1978 Hartshorn ...................... 252/187.21 4,330,531 5/1982 Alliger ................................ 424/149 73) Assignee: Rio Linda Chemical Company, Inc., Primary Examiner-Arthur L. Corbin Sacramento, Calif. Attorney, Agent, or Firm-Leonard Bloom (21) Appl. No.: 879,907 57 ABSTRACT An aqueous solution, preferably an aqueous disinfectant solution, containing chlorine dioxide and which is capa 22 Filed: Jun. 30, 1986 ble of forming a foam, and methods for the preparation and use thereof. An aqueous disinfectant solution capa Related U.S. Application Data ble of forming a foam is prepared adding a foaming agent, i.e., a suitable surfactant, to water. Chlorine diox 63 Continuation of Ser. No. 636,309, Jul. 31, 1984, aban ide may then be added to the solution or it may be doned. generated in situ by reacting an oxidizing agent, a cati onic exchange resin in the acidic form, or an acid with 51 Int. Cl.' ................................................ C11D 3/48 a metal chlorite dissolved therein. The resultant foam 52 U.S.C. -

Caring for Your Patients with Xerostomia………
CARING FOR YOUR PATIENTS WITH XEROSTOMIA……… YOU CAN MAKE A DIFFERENCE!!! Presented by: Shannon M. Nanne, RDH 1 CARING FOR YOUR PATIENTS WITH XEROSTOMIA: YOU CAN MAKE A DIFFERENCE! presented by: Shannon M. Nanne R.D.H. Introduction: This lecture will cover the causes and problems associated with xerostomia, guidelines for care, clinical observations, patient management and state of the art oral care products by Laclede, Inc. The term “xerostomia” refers to the patient’s perception of oral dryness. Xerostomia is not usually a solitary symptom. There is a wide variety of oral and non oral symptoms associated with this condition. I. Oral and Non Oral Symptoms Reported By Xerostomics A. Oral Symptoms >Halitosis / mouth odor >Desires constant moisture >Need of fluids at bedside >Difficulty with speech >Complaints of dry mouth >Difficulty with swallowing >Fissures, sores at lip corners >Sleep interruptions due to thirst >Difficulty in chewing dry food >Sips water while eating >Tongue: burning, tingling >Problems with taste >Difficulty in swallowing pills >Difficulty in wearing dentures 1. Biotene Denture Grip 2 B. Non Oral Symptoms >Dry throat >Blurred vision >Dry eyes >Burning sensations in eyes >Sandy, gritty feeling in eyes >Daily use of eye drops >Dry skin >Mouth breather >Dry nose >Change in sense of smell >Constipation >Heartburn >Vaginal itching, burning, dryness, a history of fungal infections 1. Refresh Preservative Free Eye Drops II. Functions of Saliva A. Protective Functions of Saliva Functions Salivary Components Lubrications Mucins, proline-rich proteins, water Antimicrobal Salivary proteins: lysozyme, lactoferrin, lactoperoxidase, mucins, cystatins, histatins, secretory Iga: proline-rich glycoproteins Remineralization Calcium, phosphate, statherin, anionic protein-rich proteins Cleansing Water Buffering Bicarbonate, phosphate ions Mucosal integrity Mucins, electrolytes, wate B. -

Urea-Formaldehyde Foam Insulations: a Review of Their Properties and Performance
NBS NATL INST OF STAND & TECH PUBLICATIONS i NI8T U.S. DEPARt A111D7 E^^E57 PU8UCATI0NS National Bureau of Standards NBS Technical Note 1210 Urea-Formaldehyde Foam Insulations: A Review of Their Properties and Performance Walter J, Rossiter, Jr, and Robert G, Mathey NBS NBS NBS NBS NBS NBS NBS NBS NBS NBS iS NBS NBS NBS NBS NBS NBS NBS NBS NBS NB. NBS NBS NBS NBS NBS NBS NBS NBS NBS NBS iS NBS NBS NBS NBS NBS NBS NBS NBS NBS NB. NBS NBS NBS NBS NBS NBS NBS NBS NBS NBS iS NBS NBS NBS NBS NBS NBS NBS NBS NBS NB. NBS NBS NBS NBS NBS NBS NBS NBS NBS NBS iS NBS NBS NBS NBS NBS NBS NBS NBS NBS NB. NBS NBS NBS NBS NBS NBS NBS NBS NBS NBS iS NBS NBS NBS NBS NBS NBS NBS NBS NBS NB. NBS NBS NBS National Bureau ofStandards NBS NBS iS NBS NBS NBS NBS NBS NBS NBS NBS NBS NB. NBS NBS NBS NBS NBS NBS NBS NBS NBS NBS I QC BS NBS NBS NBS NBS NBS NBS NBS NBS NB.\ ;°;„ NBS NBS NBS NBS NBS NBS NBS NBS NBS No. 1210 BS NBS NBS NBS NBS NBS NBS NBS NBS NB. 1985 c. 2 he National Bureau Tm of Standards' was established by an act of Congress on March 3, 1901. The _JL Bureau's overall goal is to strengthen and advance the nation's science and technology and facilitate their effective for application public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. -

PERSONAL CARE NORTH AMERICA Ingredients Catalog
PERSONAL CARE NORTH AMERICA Ingredients Catalog www.brenntag.com 2 BRENNTAG PERSONAL CARE Ingredients Catalog Brenntag is proud to be the global market leader in full-line ingredient and chemical distribution and your first choice for solutions to your product needs. Our company is dedicated to provide responsible distribution of raw materials and services, integrity in our business transactions, and long-lasting relationships with customers and suppliers. If you can't find what you're looking for, contact us to connect with a technical representative. TABLE OF CONTENTS Actives/Biofuctional Ingredients.................................................................................................5-8 Clays - Kaolin and Illite.....................................................................................................................9 Conditioners............................................................................................................................. 10-11 Cosmetic Powders..........................................................................................................................12 Emollients / Humectants..........................................................................................................13-16 Emulsifers..................................................................................................................................17-18 Film Formers / Transfer Resistance / Water Resistance.........................................................19-21 Lipid Lamellar Gel Systems............................................................................................................22