Microglia in Aging and Alzheimer's Disease: a Comparative Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
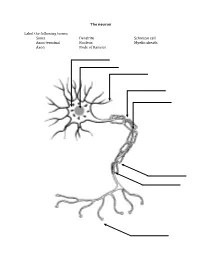
The Neuron Label the Following Terms: Soma Axon Terminal Axon Dendrite
The neuron Label the following terms: Soma Dendrite Schwaan cell Axon terminal Nucleus Myelin sheath Axon Node of Ranvier Neuron Vocabulary You must know the definitions of these terms 1. Synaptic Cleft 2. Neuron 3. Impulse 4. Sensory Neuron 5. Motor Neuron 6. Interneuron 7. Body (Soma) 8. Dendrite 9. Axon 10. Action Potential 11. Myelin Sheath (Myelin) 12. Afferent Neuron 13. Threshold 14. Neurotransmitter 15. Efferent Neurons 16. Axon Terminal 17. Stimulus 18. Refractory Period 19. Schwann 20. Nodes of Ranvier 21. Acetylcholine STEPS IN THE ACTION POTENTIAL 1. The presynaptic neuron sends neurotransmitters to postsynaptic neuron. 2. Neurotransmitters bind to receptors on the postsynaptic cell. - This action will either excite or inhibit the postsynaptic cell. - The soma becomes more positive. 3. The positive charge reaches the axon hillock. - Once the threshold of excitation is reached the neuron will fire an action potential. 4. Na+ channels open and Na+ is forced into the cell by the concentration gradient and the electrical gradient. - The neuron begins to depolarize. 5. The K+ channels open and K+ is forced out of the cell by the concentration gradient and the electrical gradient. - The neuron is depolarized. 6. The Na+ channels close at the peak of the action potential. - The neuron starts to repolarize. 7. The K+ channels close, but they close slowly and K+ leaks out. 8. The terminal buttons release neurotransmitter to the postsynaptic neuron. 9. The resting potential is overshot and the neuron falls to a -90mV (hyperpolarizes). - The neuron continues to repolarize. 10. The neuron returns to resting potential. The Synapse Label the following terms: Pre-synaptic membrane Neurotransmitters Post-synaptic membrane Synaptic cleft Vesicle Post-synaptic receptors . -

Crayfish Escape Behavior and Central Synapses
Crayfish Escape Behavior and Central Synapses. III. Electrical Junctions and Dendrite Spikes in Fast Flexor Motoneurons ROBERT S. ZUCKER Department of Biological Sciences and Program in the Neurological Sciences, Stanford University, Stanford, California 94305 THE LATERAL giant fiber is the decision fiber and fast flexor motoneurons are located in for a behavioral response in crayfish in- the dorsal neuropil of each abdominal gan- volving rapid tail flexion in response to glion. Dye injections and anatomical recon- abdominal tactile stimulation (27). Each structions of ‘identified motoneurons reveal lateral giant impulse is followed by a rapid that only those motoneurons which have tail flexion or tail flip, and when the giant dentritic branches in contact with a giant fiber does not fire in response to such stim- fiber are activated by that giant fiber (12, uli, the movement does not occur. 21). All of tl ie fast flexor motoneurons are The afferent limb of the reflex exciting excited by the ipsilateral giant; all but F4, the lateral giant has been described (28), F5 and F7 are also excited by the contra- and the habituation of the behavior has latkral giant (21 a). been explained in terms of the properties The excitation of these motoneurons, of this part of the neural circuit (29). seen as depolarizing potentials in their The properties of the neuromuscular somata, does not always reach threshold for junctions between the fast flexor motoneu- generating a spike which propagates out the rons and the phasic flexor muscles have also axon to the periphery (14). Indeed, only t,he been described (13). -

Plp-Positive Progenitor Cells Give Rise to Bergmann Glia in the Cerebellum
Citation: Cell Death and Disease (2013) 4, e546; doi:10.1038/cddis.2013.74 OPEN & 2013 Macmillan Publishers Limited All rights reserved 2041-4889/13 www.nature.com/cddis Olig2/Plp-positive progenitor cells give rise to Bergmann glia in the cerebellum S-H Chung1, F Guo2, P Jiang1, DE Pleasure2,3 and W Deng*,1,3,4 NG2 (nerve/glial antigen2)-expressing cells represent the largest population of postnatal progenitors in the central nervous system and have been classified as oligodendroglial progenitor cells, but the fate and function of these cells remain incompletely characterized. Previous studies have focused on characterizing these progenitors in the postnatal and adult subventricular zone and on analyzing the cellular and physiological properties of these cells in white and gray matter regions in the forebrain. In the present study, we examine the types of neural progeny generated by NG2 progenitors in the cerebellum by employing genetic fate mapping techniques using inducible Cre–Lox systems in vivo with two different mouse lines, the Plp-Cre-ERT2/Rosa26-EYFP and Olig2-Cre-ERT2/Rosa26-EYFP double-transgenic mice. Our data indicate that Olig2/Plp-positive NG2 cells display multipotential properties, primarily give rise to oligodendroglia but, surprisingly, also generate Bergmann glia, which are specialized glial cells in the cerebellum. The NG2 þ cells also give rise to astrocytes, but not neurons. In addition, we show that glutamate signaling is involved in distinct NG2 þ cell-fate/differentiation pathways and plays a role in the normal development of Bergmann glia. We also show an increase of cerebellar oligodendroglial lineage cells in response to hypoxic–ischemic injury, but the ability of NG2 þ cells to give rise to Bergmann glia and astrocytes remains unchanged. -

Neuregulin 1–Erbb2 Signaling Is Required for the Establishment of Radial Glia and Their Transformation Into Astrocytes in Cerebral Cortex
Neuregulin 1–erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex Ralf S. Schmid*, Barbara McGrath*, Bridget E. Berechid†, Becky Boyles*, Mark Marchionni‡, Nenad Sˇ estan†, and Eva S. Anton*§ *University of North Carolina Neuroscience Center and Department of Cell and Molecular Physiology, University of North Carolina School of Medicine, Chapel Hill, NC 27599; †Department of Neurobiology, Yale University School of Medicine, New Haven, CT 06510; and ‡CeNes Pharamceuticals, Inc., Norwood, MA 02062 Communicated by Pasko Rakic, Yale University School of Medicine, New Haven, CT, January 27, 2003 (received for review December 12, 2002) Radial glial cells and astrocytes function to support the construction mine whether NRG-1-mediated signaling is involved in radial and maintenance, respectively, of the cerebral cortex. However, the glial cell development and differentiation in the cerebral cortex. mechanisms that determine how radial glial cells are established, We show that NRG-1 signaling, involving erbB2, may act in maintained, and transformed into astrocytes in the cerebral cortex are concert with Notch signaling to exert a critical influence in the not well understood. Here, we show that neuregulin-1 (NRG-1) exerts establishment, maintenance, and appropriate transformation of a critical role in the establishment of radial glial cells. Radial glial cell radial glial cells in cerebral cortex. generation is significantly impaired in NRG mutants, and this defect can be rescued by exogenous NRG-1. Down-regulation of expression Materials and Methods and activity of erbB2, a member of the NRG-1 receptor complex, leads Clonal Analysis to Study NRG’s Role in the Initial Establishment of to the transformation of radial glial cells into astrocytes. -

Neuron Morphology Influences Axon Initial Segment Plasticity
Dartmouth College Dartmouth Digital Commons Dartmouth Scholarship Faculty Work 1-2016 Neuron Morphology Influences Axon Initial Segment Plasticity Allan T. Gulledge Dartmouth College, [email protected] Jaime J. Bravo Dartmouth College Follow this and additional works at: https://digitalcommons.dartmouth.edu/facoa Part of the Computational Neuroscience Commons, Molecular and Cellular Neuroscience Commons, and the Other Neuroscience and Neurobiology Commons Dartmouth Digital Commons Citation Gulledge, A.T. and Bravo, J.J. (2016) Neuron morphology influences axon initial segment plasticity, eNeuro doi:10.1523/ENEURO.0085-15.2016. This Article is brought to you for free and open access by the Faculty Work at Dartmouth Digital Commons. It has been accepted for inclusion in Dartmouth Scholarship by an authorized administrator of Dartmouth Digital Commons. For more information, please contact [email protected]. New Research Neuronal Excitability Neuron Morphology Influences Axon Initial Segment Plasticity1,2,3 Allan T. Gulledge1 and Jaime J. Bravo2 DOI:http://dx.doi.org/10.1523/ENEURO.0085-15.2016 1Department of Physiology and Neurobiology, Geisel School of Medicine at Dartmouth, Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire 03756, and 2Thayer School of Engineering at Dartmouth, Hanover, New Hampshire 03755 Visual Abstract In most vertebrate neurons, action potentials are initiated in the axon initial segment (AIS), a specialized region of the axon containing a high density of voltage-gated sodium and potassium channels. It has recently been proposed that neurons use plasticity of AIS length and/or location to regulate their intrinsic excitability. Here we quantify the impact of neuron morphology on AIS plasticity using computational models of simplified and realistic somatodendritic morphologies. -

Satellite Glial Cell Communication in Dorsal Root Ganglia
Neuronal somatic ATP release triggers neuron– satellite glial cell communication in dorsal root ganglia X. Zhang, Y. Chen, C. Wang, and L.-Y. M. Huang* Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston, TX 77555-1069 Edited by Charles F. Stevens, The Salk Institute for Biological Studies, La Jolla, CA, and approved April 25, 2007 (received for review December 14, 2006) It has been generally assumed that the cell body (soma) of a release of ATP from the somata of DRG neurons. Here, we show neuron, which contains the nucleus, is mainly responsible for that electric stimulation elicits robust vesicular release of ATP from synthesis of macromolecules and has a limited role in cell-to-cell neuronal somata and thus triggers bidirectional communication communication. Using sniffer patch recordings, we show here that between neurons and satellite cells. electrical stimulation of dorsal root ganglion (DRG) neurons elicits robust vesicular ATP release from their somata. The rate of release Results events increases with the frequency of nerve stimulation; external We asked first whether vesicular release of ATP occurs in the -Ca2؉ entry is required for the release. FM1–43 photoconversion somata of DRG neurons. P2X2-EGFP receptors were overex analysis further reveals that small clear vesicles participate in pressed in HEK cells and used as the biosensor (i.e., sniffer patch exocytosis. In addition, the released ATP activates P2X7 receptors method) (20). P2X2 receptors were chosen because they were easily in satellite cells that enwrap each DRG neuron and triggers the expressed at a high level (Ϸ200 pA/pF) in HEK cells and the communication between neuronal somata and glial cells. -

Myelin Biogenesis Is Associated with Pathological Ultrastructure That Is
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Myelin biogenesis is associated with pathological ultrastructure that 2 is resolved by microglia during development 3 4 5 Minou Djannatian1,2*, Ulrich Weikert3, Shima Safaiyan1,2, Christoph Wrede4, Cassandra 6 Deichsel1,2, Georg Kislinger1,2, Torben Ruhwedel3, Douglas S. Campbell5, Tjakko van Ham6, 7 Bettina Schmid2, Jan Hegermann4, Wiebke Möbius3, Martina Schifferer2,7, Mikael Simons1,2,7* 8 9 1Institute of Neuronal Cell Biology, Technical University Munich, 80802 Munich, Germany 10 2German Center for Neurodegenerative Diseases (DZNE), 81377 Munich, Germany 11 3Max-Planck Institute of Experimental Medicine, 37075 Göttingen, Germany 12 4Institute of Functional and Applied Anatomy, Research Core Unit Electron Microscopy, 13 Hannover Medical School, 30625, Hannover, Germany 14 5Department of Neuronal Remodeling, Graduate School of Pharmaceutical Sciences, Kyoto 15 University, Sakyo-ku, Kyoto 606-8501, Japan. 16 6Department of Clinical Genetics, Erasmus MC, University Medical Center Rotterdam, 3015 CN, 17 the Netherlands 18 7Munich Cluster of Systems Neurology (SyNergy), 81377 Munich, Germany 19 20 *Correspondence: [email protected] or [email protected] 21 Keywords 22 Myelination, degeneration, phagocytosis, microglia, oligodendrocytes, phosphatidylserine 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

View Software
TLR4-activated microglia have divergent effects on oligodendrocyte lineage cells DISSERTATION Presented in partial fulfillment of the requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University Evan Zachary Goldstein, BA Neuroscience Graduate Program The Ohio State University 2016 Dissertation Committee: Dr. Dana McTigue, Advisor Dr. Phillip Popovich Dr. Jonathan Godbout Dr. Courtney DeVries Copyright by Evan Zachary Goldstein 2016 i Abstract Myelin accelerates action potential conduction velocity and provides essential metabolic support for axons. Unfortunately, myelin and myelinating cells are often vulnerable to injury or disease, resulting in myelin damage, which in turn can lead to axon dysfunction, overt pathology and neurological impairment. Inflammation is a common component of CNS trauma and disease, and therefore an active inflammatory response is often considered deleterious to myelin health. While inflammation can certainly damage myelin, inflammatory processes also benefit oligodendrocyte (OL) lineage progression and myelin repair. Consistent with the divergent nature of inflammation, intraspinal toll-like receptor 4 (TLR4) activation, an innate immune pathway, kills OL lineage cells, but also initiates oligodendrogenesis. Soluble factors produced by TLR4-activated microglia can reproduce these effects in vitro, however the exact factors are unknown. To determine what microglial factors might contribute to TLR4-induced OL loss and oligodendrogenesis, mRNA of factors known to affect OL lineage cells was quantified in TLR4-activated microglia and spinal cords (chapter 2). Results indicate that TLR4-activated microglia transcribe numerous factors that induce OL loss, OL progenitor cell (OPC) proliferation and OPC differentiation. However, some factors upregulated after intraspinal TLR4 activation were not ii upregulated by microglia, suggesting that other cell types contribute to transcriptional changes in vivo. -

Tecnicas Microscopicas
CAP 1: TÉCNICAS MICROSCÓPICAS TÉCNICAS MICROSCÓPICAS 11 Lic. Carlos R. Neira Montoya Lic. Eduardo Sedano Gelvet Lic. María Elena Vilcarromero V. El estudio de los tejidos tal como los observamos hoy en día no sería posible sin la ayuda de la histotecnología; esta disciplina se encarga del estudio de los métodos técnicas y procedimientos que permiten la transformación de un órgano en una película lo suficientemente transparente y contrastada que nos permite su observación a través del microscopio (Fig. 1-1). Para que esto ocurra se tiene que seguir una serie de pasos. Cada uno de estos pasos permite la observación de las características morfológicas del tejido que nos indica la normalidad o la alteración patológica; sin embargo en estudios mucho más minuciosos, estos pasos se harán en función de las estructuras o sustancias que se deseen investigar en la muestra correspondiente. Figura 1-1. Un órgano es transformado en una película transparente. - Pág. 5 - CAP 1: TÉCNICAS MICROSCÓPICAS Los pasos de las técnicas microscópicas para obtener un preparado histológico permanente (láminas) son: 1. Toma de la muestra. 2. Fijación. 3. Inclusión. 4. Microtomía. 5. Coloración. 1. TOMA DE LA MUESTRA Es el momento que se selecciona el órgano o tejido a estudiar. De tres fuentes puede provenir el material humano: las necropsias, las biopsias y las piezas operadas. De éstas, sólo la primera puede darnos material normal; las dos últimas habitualmente proporcionarán tejidos para estudio histopatológico. - Necropsias: son las piezas que se obtienen de un cadáver. Para histología normal es necesario que se trate de un cadáver fresco y que no haya sido atacado por ninguna lesión, por lo menos el órgano que se quiere estudiar. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Improving the Gene Ontology Resource to Facilitate More Informative Analysis and Interpretation of Alzheimer’S Disease Data
G C A T T A C G G C A T genes Article Improving the Gene Ontology Resource to Facilitate More Informative Analysis and Interpretation of Alzheimer’s Disease Data Barbara Kramarz 1 , Paola Roncaglia 2 , Birgit H. M. Meldal 2 , Rachael P. Huntley 1 , Maria J. Martin 2, Sandra Orchard 2, Helen Parkinson 2, David Brough 3, Rina Bandopadhyay 4, Nigel M. Hooper 3 and Ruth C. Lovering 1,* 1 UCL Institute of Cardiovascular Science, University College London, Rayne Building, 5 University Street, London WC1E 6JF, UK; [email protected] (B.K.); [email protected] (R.P.H.) 2 European Bioinformatics Institute (EMBL-EBI), European Molecular Biology Laboratory, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK; [email protected] (P.R.); [email protected] (B.H.M.M.); [email protected] (M.J.M.); [email protected] (S.O.); [email protected] (H.P.) 3 Division of Neuroscience and Experimental Psychology, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, AV Hill Building, Oxford Road, Manchester M13 9PT, UK; [email protected] (D.B.); [email protected] (N.M.H.) 4 UCL Queen Square Institute of Neurology and Reta Lila Weston Institute of Neurological Studies, 1 Wakefield Street, London WC1N 1PJ, UK; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +44-207-679-6965 Received: 31 October 2018; Accepted: 23 November 2018; Published: 29 November 2018 Abstract: The analysis and interpretation of high-throughput datasets relies on access to high-quality bioinformatics resources, as well as processing pipelines and analysis tools. -

Copper Toxicity Links to Pathogenesis of Alzheimer's Disease And
International Journal of Molecular Sciences Review Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches Hafza Wajeeha Ejaz 1, Wei Wang 2 and Minglin Lang 1,3,* 1 CAS Center for Excellence in Biotic Interactions, College of Life Science, University of Chinese Academy of Sciences, Yuquan Road 19, Beijing 100049, China; [email protected] 2 School of Medical and Health Sciences, Edith Cowan University, Perth WA6027, Australia; [email protected] 3 College of Life Science, Agricultural University of Hebei, Baoding 071000, China * Correspondence: [email protected]; Tel.: +86-10-8825-6585/6967-2639 Received: 25 August 2020; Accepted: 29 September 2020; Published: 16 October 2020 Abstract: Alzheimer’s disease (AD) is an irreversible, age-related progressive neurological disorder, and the most common type of dementia in aged people. Neuropathological lesions of AD are neurofibrillary tangles (NFTs), and senile plaques comprise the accumulated amyloid-beta (Aβ), loaded with metal ions including Cu, Fe, or Zn. Some reports have identified metal dyshomeostasis as a neurotoxic factor of AD, among which Cu ions seem to be a central cationic metal in the formation of plaque and soluble oligomers, and have an essential role in the AD pathology. Cu-Aβ complex catalyzes the generation of reactive oxygen species (ROS) and results in oxidative damage. Several studies have indicated that oxidative stress plays a crucial role in the pathogenesis of AD. The connection of copper levels in AD is still ambiguous, as some researches indicate a Cu deficiency, while others show its higher content in AD, and therefore there is a need to increase and decrease its levels in animal models, respectively, to study which one is the cause.