The Importance of Strain (Preorganization) in Beryllium Bonds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![And [Z–Hali]- Halogen Bonds: Electron Density Properties And](https://docslib.b-cdn.net/cover/6945/and-z-hali-halogen-bonds-electron-density-properties-and-166945.webp)
And [Z–Hali]- Halogen Bonds: Electron Density Properties And
molecules Article Strength of the [Z–I···Hal]− and [Z–Hal···I]− Halogen Bonds: Electron Density Properties and Halogen Bond Length as Estimators of Interaction Energy Maxim L. Kuznetsov 1,2 1 Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Avenida Rovisco Pais, 1049-001 Lisbon, Portugal; [email protected]; Tel.: +351-218-419-236 2 Institute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, 199034 Saint Petersburg, Russia Abstract: Bond energy is the main characteristic of chemical bonds in general and of non-covalent interactions in particular. Simple methods of express estimates of the interaction energy, Eint, us- ing relationships between Eint and a property which is easily accessible from experiment is of great importance for the characterization of non-covalent interactions. In this work, practically important relationships between Eint and electron density, its Laplacian, curvature, potential, kinetic, and total energy densities at the bond critical point as well as bond length were derived for the structures of the [Z–I···Hal]− and [Z–Hal···I]− types bearing halogen bonds and involving iodine as interact- ing atom(s) (totally 412 structures). The mean absolute deviations for the correlations found were 2.06–4.76 kcal/mol. Keywords: bond critical point properties; interaction energy; bond energy; bond strength; den- sity functional theory; electron density; energy density; halogen bond; QTAIM Citation: Kuznetsov, M.L. Strength of the [Z–I···Hal]− and [Z–Hal···I]− Halogen Bonds: Electron Density Properties and Halogen Bond Length as Estimators of Interaction Energy. 1. Introduction Molecules 2021, 26, 2083. https:// The halogen bond is one of the most important types of non-covalent interactions doi.org/10.3390/molecules26072083 being second only to hydrogen bonds in its significance. -

Hypochlorous Acid Handling
Hypochlorous Acid Handling 1 Identification of Petitioned Substance 2 Chemical Names: Hypochlorous acid, CAS Numbers: 7790-92-3 3 hypochloric(I) acid, chloranol, 4 hydroxidochlorine 10 Other Codes: European Community 11 Number-22757, IUPAC-Hypochlorous acid 5 Other Name: Hydrogen hypochlorite, 6 Chlorine hydroxide List other codes: PubChem CID 24341 7 Trade Names: Bleach, Sodium hypochlorite, InChI Key: QWPPOHNGKGFGJK- 8 Calcium hypochlorite, Sterilox, hypochlorite, UHFFFAOYSA-N 9 NVC-10 UNII: 712K4CDC10 12 Summary of Petitioned Use 13 A petition has been received from a stakeholder requesting that hypochlorous acid (also referred 14 to as electrolyzed water (EW)) be added to the list of synthetic substances allowed for use in 15 organic production and handling (7 CFR §§ 205.600-606). Specifically, the petition concerns the 16 formation of hypochlorous acid at the anode of an electrolysis apparatus designed for its 17 production from a brine solution. This active ingredient is aqueous hypochlorous acid which acts 18 as an oxidizing agent. The petitioner plans use hypochlorous acid as a sanitizer and antimicrobial 19 agent for the production and handling of organic products. The petition also requests to resolve a 20 difference in interpretation of allowed substances for chlorine materials on the National List of 21 Allowed and Prohibited Substances that contain the active ingredient hypochlorous acid (NOP- 22 PM 14-3 Electrolyzed water). 23 The NOP has issued NOP 5026 “Guidance, the use of Chlorine Materials in Organic Production 24 and Handling.” This guidance document clarifies the use of chlorine materials in organic 25 production and handling to align the National List with the November, 1995 NOSB 26 recommendation on chlorine materials which read: 27 “Allowed for disinfecting and sanitizing food contact surfaces. -
Proceedings of the Indiana Academy of Science
A Comparison of Halogeno and Oxyacids L53 A COMPARISON OF HALOGENO AND OXYACIDS J. A. Nieuwland and T. H. Vaughn, University of Notre Dame In a recent paper originating in this department and sent to one of the chemical journals for publication the term "fluo acid" was used to designate a flourine containing acid which could be considered as having been formed by substituting two flourine atoms for each oxygen atom in an oxyacid. Criticism of this term on the ground that it was novel and unusual resulted. The paper mentioned being essentially organic in its nature it was impossible to explain the matter sufficiently except as a separate discussion. The purpose of this paper is to make an attempt to establish the term "halogeno acid" as a general name for acids in which the halogens may be considered to replace the oxygen in oxyacids and to establish the terms, fluo, chloro, bromo, and iodo as the specific names of these acids where the halogen involved is flourine, chlorine, bromine, and iodine respectively. As a matter of fact these names have been used in several cases and are accepted terms for those cases, i.e., fluoboric acid, bromostannic acid, fluoplumbic acid, fluosilicic acid, etc. When various molecular portions of water are added to the anhydrides of the acid forming elements, the several oxyacids are produced. B 2 3 +H 2 0^2HB0 2 B 2 2 +3H 2 0->2H 3 B0 3 In an analogous manner when the halogen acids are added to the halogen compounds corresponding to the anhydrides of the acid forming elements, the halogeno acids are formed. -

Atoms, Molecules, Ions, and Inorganic Nomenclature
Atoms, Molecules, Ions, and Inorganic Nomenclature Brown, LeMay Ch 2 AP Chemistry Monta Vista High School 2.2: Evidence for the Atomic Theory 1. J.J. Thomson’s cathode ray tube: discovery of electrons and the e- charge-to-mass ratio ¶ In a vacuum chamber, flow of high voltage (emitted from cathode to anode) is deflected by magnetic & electrical fields (animation: http://highered.mcgraw-hill.com/olcweb/cgi/pluginpop.cgi?it=swf::100%::100%::/sites/dl/free/ 0072512644/117354/01_Cathode_Ray_Tube.swf::Cathode%20Ray%20Tube) 2 2. Robert Millikan’s oil drop: determines charge of e- (and thus the mass) ¶ “Atomized” drops of oil picked up small charges (integral numbers), and balanced oil drops in an electrical & gravitational field http://cwx.prenhall.com/petrucci/medialib/media_portfolio/text_images/004_MILLIKANOIL.MOV 3. Ernest Rutherford’s gold foil: discovery of nucleus as center of positive charge ¶ Alpha particles from radioactive source are deflected from positive gold atom nuclei http://www.mhhe.com/physsci/ chemistry/animations/ chang_2e/ rutherfords_experiment.swf 2.3: Structure of the Atom Figure 1: Subatomic particles (Table 2.1; 1 amu = 1.66054 x 10-24 g). Subatomic Charge Location Mass particle Proton, p+ +1.6 x 10-19 C nucleus 1.0073 amu Neutron, n None nucleus 1.0087 amu Electron, e- -1.6 x 10-19 C e- cloud 5.486 x 10-4 amu 5 Vocabulary n Atomic number: number of p+ (determines the element) n Mass number: sum of p+ and n (determines the isotope) n Isotopes: atoms of an element that differ in the number of neutrons n Isobars: atoms of different elements with same atomic mass but different atomic number. -

A New Way for Probing Bond Strength J
A New Way for Probing Bond Strength J. Klein, H. Khartabil, J.C. Boisson, J. Contreras-Garcia, Jean-Philip Piquemal, E. Henon To cite this version: J. Klein, H. Khartabil, J.C. Boisson, J. Contreras-Garcia, Jean-Philip Piquemal, et al.. A New Way for Probing Bond Strength. Journal of Physical Chemistry A, American Chemical Society, 2020, 124 (9), pp.1850-1860. 10.1021/acs.jpca.9b09845. hal-02377737 HAL Id: hal-02377737 https://hal.archives-ouvertes.fr/hal-02377737 Submitted on 27 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. A New Way for Probing Bond Strength Johanna Klein,y Hassan Khartabil,y Jean-Charles Boisson,z Julia Contreras-Garc´ıa,{ Jean-Philip Piquemal,{ and Eric H´enon∗,y yInstitut de Chimie Mol´eculaire de Reims UMR CNRS 7312, Universit´ede Reims Champagne-Ardenne, Moulin de la Housse 51687 Reims Cedex 02 BP39 (France) zCReSTIC EA 3804, Universit´ede Reims Champagne-Ardenne, Moulin de la Housse 51687 Reims Cedex 02 BP39 (France) {Sorbonne Universit´es,UPMC, Laboratoire de Chimie Th´eoriqueand UMR CNRS 7616, 4 Pl Jussieu, 75252 Paris Cedex 05(France) E-mail: [email protected] Phone: +33(3)26918497 1 Abstract The covalent chemical bond is intimately linked to electron sharing between atoms. -

Humidity-Sensing Component Composition
Patentamt JEuropaischesEuropean Patent Office © Publication number: 0 242 834 Office europeen des brevets A2 © EUROPEAN PATENT APPLICATION © Application number: 87105802.0 © Int.CI.3: G 01 N 27/12 @ Dateoffiling: 21.04.87 © Priority: 24.04.86 JP 93211/86 ©Applicant: MITSUBISHI GAS CHEMICAL COMPANY, INC. 24.04.86 JP 93212/86 5-2,Marunouchi2-chomeChiyoda-Ku 23.12.86 JP 305392/86 Tokyo(JP) © Inventor: Sugio, Akitoshi © Dateof publication of application: 382-155, Besho Ohaza-Sashiougiryo 28.10.87 Bulletin 87/44 Ohmiya-shiSaitama-Ken(JP) © Designated Contracting States: © Inventor: Shimomura.Tadashi FR GB 11 05-21, Higashifukai Nagareyama-shi Chiba-Ken(JP) @ Inventor: Wakabayashi, Hidechika 6-101, Nishlkubocho 15-chome Tokiwadaira Matsudo-shl Chiba-Ken(JP) © Inventor: Kondo,Osamu 25-15-201, Niijyuku 5-chome Katsushika-ku Tokyo(JP) @ Inventor: Ogasawara, Kazuharu 1 1 -1 6, Kanamachi 5-chome Katsushika-ku Tokyo(JP) © Inventor: Nishizawa, Chiharu 17-1,Kanegasaku Matsudo-shl Chiba-Ken(JP) © Representative: Blumbach Weser Bergen Kramer Zwirner Hoffmann Patentanwalte Radeckestrasse43 D-8000Munchen60(DE) Humidity-sensing component composition. © A humidity-sensing component composition includes a metallic oxide and a chalcogen oxyacid salt represented by a general formula AxByOz where A is one of an alkali metal and an alkaline earth metal, B is one of sulphur, selenium, and tellurium, O is oxygen, x is 1 to 2, y 1 to 5, and z 2 to 7. The chalcogen oxyacid salt is blended by an amount of 0.01 to 99.99 mol% in the metallic oxide with the sum of the metallic oxide and the chalcogen oxyacid salt as a reference. -

Halogen Bonds in Biological Molecules
Halogen bonds in biological molecules Pascal Auffinger†‡, Franklin A. Hays§, Eric Westhof†, and P. Shing Ho‡§ †Institut de Biologie Mole´culaire et Cellulaire, Centre National de la Recherche Scientifique, Unite´Propre de Recherche 9002, Universite´Louis Pasteur, 15 Rue Rene´Descartes, F-67084 Strasbourg, France; and §Department of Biochemistry and Biophysics, Agriculture͞Life Sciences Building, Room 2011, Oregon State University, Corvallis, OR 97331-7305 Communicated by K. E. van Holde, Oregon State University, Corvallis, OR, October 13, 2004 (received for review August 17, 2004) Short oxygen–halogen interactions have been known in organic chemistry since the 1950s and recently have been exploited in the design of supramolecular assemblies. The present survey of protein and nucleic acid structures reveals similar halogen bonds as po- tentially stabilizing inter- and intramolecular interactions that can affect ligand binding and molecular folding. A halogen bond in biomolecules can be defined as a short COX⅐⅐⅐OOY interaction (COX is a carbon-bonded chlorine, bromine, or iodine, and OOY is a carbonyl, hydroxyl, charged carboxylate, or phosphate group), where the X⅐⅐⅐O distance is less than or equal to the sums of the respective van der Waals radii (3.27 Å for Cl⅐⅐⅐O, 3.37Å for Br⅐⅐⅐O, and 3.50 Å for I⅐⅐⅐O) and can conform to the geometry seen in small molecules, with the COX⅐⅐⅐O angle Ϸ165° (consistent with a strong Fig. 1. Schematic of short halogen (X) interactions to various oxygen- directional polarization of the halogen) and the X⅐⅐⅐OOY angle containing functional groups (where OOY can be a carbonyl, hydroxyl, or Ϸ120°. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Binuclear Copper(I) Borohydride Complex Containing Bridging Bis
crystals Article Binuclear Copper(I) Borohydride Complex Containing Bridging Bis(diphenylphosphino) Methane Ligands: Polymorphic Structures of 2 [(µ2-dppm)2Cu2(η -BH4)2] Dichloromethane Solvate Natalia V. Belkova 1 ID , Igor E. Golub 1,2 ID , Evgenii I. Gutsul 1, Konstantin A. Lyssenko 1, Alexander S. Peregudov 1, Viktor D. Makhaev 3, Oleg A. Filippov 1 ID , Lina M. Epstein 1, Andrea Rossin 4 ID , Maurizio Peruzzini 4 and Elena S. Shubina 1,* ID 1 A. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences (INEOS RAS), 119991 Moscow, Russia; [email protected] (N.V.B.); [email protected] (I.E.G.); [email protected] (E.I.G.); [email protected] (K.A.L.); [email protected] (A.S.P.); [email protected] (O.A.F.); [email protected] (L.M.E.) 2 Inorganic Chemistry Department, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia 3 Institute of Problems of Chemical Physics, Russian Academy of Sciences (IPCP RAS), 142432 Moscow, Russia; [email protected] 4 Istituto di Chimica dei Composti Organometallici Consiglio Nazionale delle Ricerche (ICCOM CNR), 50019 Sesto Fiorentino, Italy; [email protected] (A.R.); [email protected] (M.P.) * Correspondence: [email protected]; Tel.: +7-495-135-5085 Academic Editor: Sławomir J. Grabowski Received: 18 September 2017; Accepted: 17 October 2017; Published: 20 October 2017 Abstract: Bis(diphenylphosphino)methane copper(I) tetrahydroborate was synthesized by ligands exchange in bis(triphenylphosphine) copper(I) tetrahydroborate, and characterized by XRD, FTIR, NMR spectroscopy. According to XRD the title compound has dimeric structure, [(µ2-dppm)2Cu2(η2-BH4)2], and crystallizes as CH2Cl2 solvate in two polymorphic forms (orthorhombic, 1, and monoclinic, 2) The details of molecular geometry and the crystal-packing pattern in polymorphs were studied. -
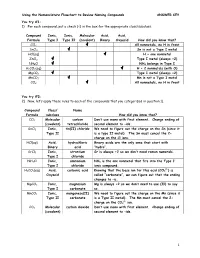
Using the Nomenclature Flowchart to Review Naming Compounds ANSWER KEY
Using the Nomenclature Flowchart to Review Naming Compounds ANSWER KEY You try #1: 1) For each compound, put a check () in the box for the appropriate class/subclass. Compound Ionic, Ionic, Molecular Acid, Acid, Formula Type I Type II (covalent) Binary Oxyacid How did you know that? CCl4 All nonmetals, no H in front SnCl2 Sn is not a Type I metal HCl(aq) H + one nonmetal ZnCl2 Type I metal (always +2) NH4Cl NH4 belongs in Type I H2CO3(aq) H + 2 nonmetals (with O) MgCO3 Type I metal (always +2) MnCO3 Mn is not a Type I metal CO2 All nonmetals, no H in front You try #2: 2) Now, let’s apply these rules to each of the compounds that you categorized in question 1). Compound Class/ Name Formula subclass How did you know that? CCl4 Molecular carbon Don’t use mono with first element. Change ending of (covalent) tetrachloride second element to –ide. SnCl2 Ionic, tin(II) chloride We need to figure out the charge on the Sn (since it Type II is a type II metal). The Sn must cancel the 2- charge on the Cl ions. HCl(aq) Acid, hydrochloric Binary acids are the only ones that start with Binary acid “hydro”. SrCl2 Ionic, strontium Sr is always +2 so we don’t need roman numerals. Type I chloride NH4Cl Ionic, ammonium NH4 is the one nonmetal that fits into the Type I Type I chloride ionic compound. 2- H2CO3(aq) Acid, carbonic acid Knowing that the base ion for this acid (CO3 ) is Oxyacid called “carbonate”, we can figure out that the ending changes to –ic. -

Selective Synthesis of Manganese/Silicon Complexes in Supercritical Water
Hindawi Publishing Corporation Journal of Nanomaterials Volume 2014, Article ID 713685, 8 pages http://dx.doi.org/10.1155/2014/713685 Research Article Selective Synthesis of Manganese/Silicon Complexes in Supercritical Water Jiancheng Wang,1 Zhaoliang Peng,1 Bing Wang,1 Lina Han,1,2 Liping Chang,1 Weiren Bao,1 and Gang Feng3 1 State Key Laboratory Breeding Base of Coal Science and Technology Co-founded by Shanxi Province and the Ministry of Science and Technology, Taiyuan University of Technology, Taiyuan 030024, China 2 College of Materials Science and Engineering, Taiyuan University of Technology, Taiyuan 030024, China 3 Shanghai Research Institute of Petrochemical Technology, SINOPEC, Shanghai 201208, China Correspondence should be addressed to Weiren Bao; [email protected] and Gang Feng; [email protected] Received 9 February 2014; Accepted 14 March 2014; Published 4 May 2014 Academic Editor: Wen Zeng Copyright © 2014 Jiancheng Wang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Aseriesofmanganesesalts(Mn(NO3)2, MnCl2, MnSO4,andMn(Ac)2) and silicon materials (silica sand, silica sol, and tetraethyl orthosilicate) were used to synthesize Mn/Si complexes in supercritical water using a tube reactor. X-ray diffraction (XRD), X- ray photoelectron spectrometer (XPS), transmission electron microscopy (TEM), and scanning electron microscopy (SEM) were employed to characterize the structure and morphology of the solid products. It was found that MnO2,Mn2O3,andMn2SiO4 could be obtained in supercritical water at 673 K in 5 minutes. -

Theoretical Study on the Noncovalent Interactions Involving Triplet Diphenylcarbene
Theoretical Study on the Noncovalent Interactions Involving Triplet Diphenylcarbene Chunhong Zhao Hebei Normal University Huihua College Hui Lin Hebei Normal University Aiting Shan Hebei Normal University Shaofu Guo Hebei Normal University Huihua College Xiaoyan Li Hebei Normal University Xueying Zhang ( [email protected] ) Hebei Normal University https://orcid.org/0000-0003-1598-8501 Research Article Keywords: triplet diphenylcarbene, noncovalent interaction, electron density shift, electron spin density Posted Date: May 19th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-445417/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Journal of Molecular Modeling on July 9th, 2021. See the published version at https://doi.org/10.1007/s00894-021-04838-6. Page 1/21 Abstract The properties of some types of noncovalent interactions formed by triplet diphenylcarbene (DPC3) have been investigated by means of density functional theory (DFT) calculations and quantum theory of 3 atoms in molecules (QTAIM) studies. The DPC ···LA (LA = AlF3, SiF4, PF5, SF2, ClF) complexes have been analyzed from their equilibrium geometries, binding energies, charge transfer and properties of electron 3 density. The triel bond in the DPC ···AlF3 complex exhibits a partially covalent nature, with the binding energy − 65.7kJ/mol. The tetrel bond, pnicogen bond, chalcogen bond and halogen bond in the DPC3···LA (LA = SiF4, PF5, SF2, ClF) complexes show the character of a weak closed-shell noncovalent interaction. Polarization plays an important role in the formation of the studied complexes.