Integrative Analysis of Oncogenic Fusion Genes and Their Functional Impact in Colorectal Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Universidade Estadual De Campinas Instituto De Biologia
UNIVERSIDADE ESTADUAL DE CAMPINAS INSTITUTO DE BIOLOGIA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA CAMPINAS 2016 1 VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA Dissertação apresentada ao Instituto de Biologia da Universidade Estadual de Campinas como parte dos requisitos exigidos para a obtenção do Título de Mestra em Biologia Funcional e Molecular na área de concentração de Bioquímica. Dissertation presented to the Institute of Biology of the University of Campinas in partial fulfillment of the requirements for the degree of Master in Functional and Molecular Biology, in the area of Biochemistry. ESTE ARQUIVO DIGITAL CORRESPONDE À VERSÃO FINAL DA DISSERTAÇÃO DEFENDIDA PELA ALUNA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA E ORIENTADA PELO DANIEL MARTINS-DE-SOUZA. Orientador: Daniel Martins-de-Souza CAMPINAS 2016 2 Agência(s) de fomento e nº(s) de processo(s): CNPq, 151787/2F2014-0 Ficha catalográfica Universidade Estadual de Campinas Biblioteca do Instituto de Biologia Mara Janaina de Oliveira - CRB 8/6972 Saia-Cereda, Verônica Aparecida Monteiro, 1988- Sa21p O proteoma do corpo caloso da esquizofrenia / Verônica Aparecida Monteiro Saia Cereda. – Campinas, SP : [s.n.], 2016. Orientador: Daniel Martins de Souza. Dissertação (mestrado) – Universidade Estadual de Campinas, Instituto de Biologia. 1. Esquizofrenia. 2. Espectrometria de massas. 3. Corpo caloso. -

N-Glycan Trimming in the ER and Calnexin/Calreticulin Cycle
Neurotransmitter receptorsGABA and A postsynapticreceptor activation signal transmission Ligand-gated ion channel transport GABAGABA Areceptor receptor alpha-5 alpha-1/beta-1/gamma-2 subunit GABA A receptor alpha-2/beta-2/gamma-2GABA receptor alpha-4 subunit GABAGABA receptor A receptor beta-3 subunitalpha-6/beta-2/gamma-2 GABA-AGABA receptor; A receptor alpha-1/beta-2/gamma-2GABA receptoralpha-3/beta-2/gamma-2 alpha-3 subunit GABA-A GABAreceptor; receptor benzodiazepine alpha-6 subunit site GABA-AGABA-A receptor; receptor; GABA-A anion site channel (alpha1/beta2 interface) GABA-A receptor;GABA alpha-6/beta-3/gamma-2 receptor beta-2 subunit GABAGABA receptorGABA-A receptor alpha-2receptor; alpha-1 subunit agonist subunit GABA site Serotonin 3a (5-HT3a) receptor GABA receptorGABA-C rho-1 subunitreceptor GlycineSerotonin receptor subunit3 (5-HT3) alpha-1 receptor GABA receptor rho-2 subunit GlycineGlycine receptor receptor subunit subunit alpha-2 alpha-3 Ca2+ activated K+ channels Metabolism of ingested SeMet, Sec, MeSec into H2Se SmallIntermediateSmall conductance conductance conductance calcium-activated calcium-activated calcium-activated potassium potassium potassiumchannel channel protein channel protein 2 protein 1 4 Small conductance calcium-activatedCalcium-activated potassium potassium channel alpha/beta channel 1 protein 3 Calcium-activated potassiumHistamine channel subunit alpha-1 N-methyltransferase Neuraminidase Pyrimidine biosynthesis Nicotinamide N-methyltransferase Adenosylhomocysteinase PolymerasePolymeraseHistidine basic -

Regulation of Skeletal Muscle Metabolism and Development by Small, Non-Coding Rnas: Implications for Insulin Resistance and Type 2 Diabetes Mellitus
From the Department of Molecular Medicine and Surgery Karolinska Institutet, Stockholm, Sweden Regulation of Skeletal Muscle Metabolism and Development by Small, Non-Coding RNAs: Implications for Insulin Resistance and Type 2 Diabetes Mellitus Rasmus Sjögren Stockholm 2018 All previously published papers were reproduced with permission from the publisher. © 2017 by the American Diabetes Association ® Diabetes 2017 Jul; 66(7): 1807-1818 Reprinted with permission from the American Diabetes Association ® Published by Karolinska Institutet. Printed by Eprint AB, 2018. © Rasmus Sjögren, 2018. ISBN 978-91-7831-057-9 Department of Molecular Medicine and Surgery Regulation of Skeletal Muscle Metabolism and Development by Small, Non-Coding RNAs: Implications for Insulin Resistance and Type 2 Diabetes Mellitus THESIS FOR DOCTORAL DEGREE (Ph.D.) by Rasmus Sjögren Defended on: Wednesday the 13th of June 2018, 12:30 pm Hillarpsalen, Retzius väg 8, Stockholm Principal Supervisor: Opponent: Prof Juleen R. Zierath Prof Markus Stoffel Karolinska Institutet Swiss Federal Institute of Technology in Zurich Department of Molecular Medicine and Surgery Department of Biology Division of Integrative Physiology Division of Metabolism and Metabolic Diseases Co-supervisor(s): Examination Board: Prof Anna Krook Prof Ulf Eriksson Karolinska Institutet Karolinska Insitutet Department of Physiology and Pharmacology Department of Medical Biochemistry and Bio- Division of Integrative Physiology physics Division of Vascular Biology Prof Eckardt Treuter Karolinska Insitutet Department of Biosciences and Nutrition Division of Epigenome Regulation Prof Lena Eliasson Lund University Department of Clinical Sciences, Malmö Division of Islet Cell Exocytosis ABSTRACT microRNAs (miRNAs) are a class of epigenetic post-transcriptional regulators. These short (~22 nucleotides) non-coding RNAs can potently reduce protein abundance through direction of the RNA-induced silencing complex to targeted genes. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Transdifferentiation of Chicken Embryonic Cells Into Muscle Cells by the 3' Untranslated Region of Muscle Tropomyosin THOMAS J
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 7520-7524, August 1995 Cell Biology Transdifferentiation of chicken embryonic cells into muscle cells by the 3' untranslated region of muscle tropomyosin THOMAS J. L'ECUYER*, PAUL C. TOMPACHt, ERIC MORRISt, AND ALICE B. FULTON*§ Departments of tBiochemistry, tOral and Maxillofacial Surgery, and *Pediatrics, University of Iowa, Iowa City, IA 52242 Communicated by Sheldon Penman, Massachusetts Institute of Technology, Cambridge, MA, May 3, 1995 ABSTRACT Transfection with a plasmid encoding the 3' Drosophila pattern formation is influenced by the 3' UTRs untranslated region (3' UTR) of skeletal muscle tropomyosin of nanos (23) and bicoid (24). The posterior determinant induces chicken embryonic fibroblasts to express skeletal nanos, important in abdominal segmentation, exerts its effect tropomyosin. Such cells become spindle shaped, fuse, and by its 3' UTR inhibiting maternal hb gene expression. Dro- express titin, a marker of striated muscle differentiation. sophila embryos acquire anterior-position determination by a Skeletal muscle tropomyosin and titin organize in sarcomeric gradient of bicoid protein; this gradient requires the 3' UTR arrays. When the tropomyosin 3' UTR is expressed in osteo- of bicoid mRNA. blasts, less skeletal muscle tropomyosin is expressed, and titin A 3' UTR can affect mineral metabolism. Eukaryotic sel- expression is delayed. Some transfected osteoblasts become enoproteins have sites within the 3' UTR that are critical for spindle shaped but do not fuse nor organize these proteins into selenocysteine incorporation in the coding region (25). An sarcomeres. Transfected cells expressing muscle tropomyosin iron response element within the ferritin and transferrin organize muscle and nonmuscle isoforms into the same struc- receptor transcripts allows the expression of these genes to be tures. -

Intrinsic Indicators for Specimen Degradation
Laboratory Investigation (2013) 93, 242–253 & 2013 USCAP, Inc All rights reserved 0023-6837/13 $32.00 Intrinsic indicators for specimen degradation Jie Li1, Catherine Kil1, Kelly Considine1, Bartosz Smarkucki1, Michael C Stankewich1, Brian Balgley2 and Alexander O Vortmeyer1 Variable degrees of molecular degradation occur in human surgical specimens before clinical examination and severely affect analytical results. We therefore initiated an investigation to identify protein markers for tissue degradation assessment. We exposed 4 cell lines and 64 surgical/autopsy specimens to defined periods of time at room temperature before procurement (experimental cold ischemic time (CIT)-dependent tissue degradation model). Using two-dimen- sional fluorescence difference gel electrophoresis in conjunction with mass spectrometry, we performed comparative proteomic analyses on cells at different CIT exposures and identified proteins with CIT-dependent changes. The results were validated by testing clinical specimens with western blot analysis. We identified 26 proteins that underwent dynamic changes (characterized by continuous quantitative changes, isoelectric changes, and/or proteolytic cleavages) in our degradation model. These changes are strongly associated with the length of CIT. We demonstrate these proteins to represent universal tissue degradation indicators (TDIs) in clinical specimens. We also devised and implemented a unique degradation measure by calculating the quantitative ratio between TDIs’ intact forms and their respective degradation- -
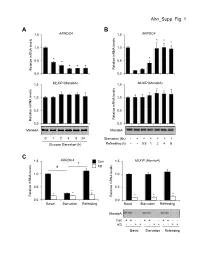
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

MAPK4 Overexpression Promotes Tumor Progression Via Noncanonical Activation of AKT/Mtor Signaling
The Journal of Clinical Investigation RESEARCH ARTICLE MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling Wei Wang,1 Tao Shen,1 Bingning Dong,1 Chad J. Creighton,2,3 Yanling Meng,1 Wolong Zhou,1 Qing Shi,1 Hao Zhou,1 Yinjie Zhang,1 David D. Moore,1 and Feng Yang1 1Department of Molecular and Cellular Biology, 2Department of Medicine, and 3Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, Texas, USA. MAPK4 is an atypical MAPK. Currently, little is known about its physiological function and involvement in diseases, including cancer. A comprehensive analysis of 8887 gene expression profiles in The Cancer Genome Atlas (TCGA) revealed that MAPK4 overexpression correlates with decreased overall survival, with particularly marked survival effects in patients with lung adenocarcinoma, bladder cancer, low-grade glioma, and thyroid carcinoma. Interestingly, human tumor MAPK4 overexpression also correlated with phosphorylation of AKT, 4E-BP1, and p70S6K, independent of the loss of PTEN or mutation of PIK3CA. This led us to examine whether MAPK4 activates the key metabolic, prosurvival, and proliferative kinase AKT and mTORC1 signaling, independent of the canonical PI3K pathway. We found that MAPK4 activated AKT via a novel, concerted mechanism independent of PI3K. Mechanistically, MAPK4 directly bound and activated AKT by phosphorylation of the activation loop at threonine 308. It also activated mTORC2 to phosphorylate AKT at serine 473 for full activation. MAPK4 overexpression induced oncogenic outcomes, including transforming prostate epithelial cells into anchorage-independent growth, and MAPK4 knockdown inhibited cancer cell proliferation, anchorage-independent growth, and xenograft growth. We concluded that MAPK4 can promote cancer by activating the AKT/mTOR signaling pathway and that targeting MAPK4 may provide a novel therapeutic approach for cancer. -

S1 Table Protein
S1 Table dFSHD12_TE dFSHD12_NE aFSHD51_TE aFSHD51_NE Accession Gene Protein H/L SD # bold H/L SD # bold H/L SD # bold H/L SD # bold Intermediate filament (or associated proteins) P17661 DES Desmin 0.91 N.D. 37 1.06 1.35 19 0.89 1,13 33 * 1.20 1.21 17 * P02545 LMNA Prelamin-A/C 0.90 1.25 30 * 1.07 1.26 20 0.96 1.23 34 1.21 1.24 19 * P48681 NES Nestin 0.91 N.D. 60 0.94 1.25 27 0.72 1.20 50 * 0.89 1.27 31 * P08670 VIM Vimentin 1.04 N.D. 35 1.24 1.24 14 * 1.21 1.18 36 * 1.39 1.17 16 * Q15149 PLEC Plectin-1 1.10 1.28 19 1.05 1.10 3 1.07 1.23 26 1.25 1.27 7 * P02511 CRYAB Alpha-crystallin B chain (HspB5) 1.47 1.17 2 1.17 2 1.14 1.12 2 Tubulin (or associated proteins) P62158 CALM1 Calmodulin (CaM) 0.83 0.00 1 0.93 0.00 1 Programmed cell death 6- Q8WUM4 PDCD6IP 1.34 0.00 1 interacting protein Q71U36 TUBA1A Tubulin α-1A chain (α-tubulin 3) 1.44 1.12 3 * Tubulin β chain (Tubulin β-5 P07437 TUBB 1.52 0.00 1 chain) Nuclear mitotic apparatus Q14980 NUMA1 0.68 0.00 1 0.75 0.00 1 protein 1 Microtubule-associated protein P27816 MAP4 1.10 1 4 Microtubule-associated protein P78559 MAP1A 0.76 0.00 1 1A Q6PEY2 TUBA3E Tubulin α-3E chain (α-tubulin 3E) 0.91 0.00 1 1.12 0.00 1 0.98 1.12 3 Tubulin β-2C chain (Tubulin β-2 P68371 TUBB2C 0.93 1.13 7 chain) Q3ZCM7 TUBB8 Tubulin β-8 chain 0.97 1.09 4 Serine P34897 SHMT2 hydroxymethyltransferase 0.93 0.00 1 (serine methylase) Cytoskeleton-associated protein Q07065 CKAP4 0.98 1.24 5 0.96 0.00 1 1.08 1.28 6 0.75 0.00 1 4 (p63) Centrosomal protein of 135 kDa Q66GS9 CEP135 0.84 0.00 1 (Centrosomal protein 4) Pre-B cell leukemia transcription Q96AQ6 PBXIP1 0.74 0.00 1 0.80 1 factor-interacting protein 1 T-complex protein 1 subunit P17897 TCP1 0.84 0.00 1 alpha (CCT-alpha) Cytoplasmic dynein Q13409 DYNC1I2 1.01 0.00 1 intermediate chain 2 (DH IC-2) Dynein heavy chain 3 (Dnahc3- Q8TD57 DNAH3 b) Microtubule-actin cross- linking Q9UPN3 MACF1 factor 1 (Trabeculin-alpha) Actin (or associated including myofibril-associated porteins) P60709 ACTB Actin, cytoplasmic 1 (β-actin) 1.11 N.D. -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -
Probe Set Gene Title Gene Symbol Fold Change Down/Up P
Supplementary Table 1. Regulated genes in mice skeletal muscle after DHT and E2 Probe Set Gene Title Gene Symbol Fold Change Down/Up P Genes regulated after DHT 1419487_at myosin binding protein H Mybph 11.9 down 0.000 1415927_at Actin. alpha. cardiac Actc1 3.8 down 0.000 1419606_a_at troponin T1. skeletal. slow Tnnt1 2.8 down 0.004 1448553_at myosin. heavy polypeptide 7. cardiac muscle. beta Myh7 2.6 down 0.001 1417765_a_at amylase 1. salivary Amy1 2.5 down 0.000 1416411_at glutathione S-transferase. mu 2 Gstm2 2.5 down 0.000 1418492_at gremlin 2 homolog. cysteine knot superfamily (Xenopus laevis) Grem2 2.4 down 0.000 1424392_at alcohol dehydrogenase. iron containing. 1 Adhfe1 2.2 down 0.000 1418370_at troponin C. cardiac/slow skeletal Tnnc1 2.2 down 0.002 1448394_at myosin. light polypeptide 2. regulatory. cardiac. slow Myl2 2.2 down 0.004 1450813_a_at troponin I. skeletal. slow 1 Tnni1 2.1 down 0.001 1449996_a_at tropomyosin 3. gamma Tpm3 2.1 down 0.017 1449118_at dihydrolipoamide branched chain transacylase E2 Dbt 1.8 down 0.002 1418507_s_at suppressor of cytokine signaling 2 Socs2 1.8 down 0.038 1449059_a_at 3-oxoacid CoA transferase 1 Oxct1 1.8 down 0.000 1453552_at RIKEN cDNA 2310014F07 gene 2310014F07Rik 1.8 down 0.012 1436913_at CDC14 cell division cycle 14 homolog A (S. cerevisiae) Cdc14a 1.8 down 0.014 1448554_s_at myosin. heavy polypeptide 6. cardiac muscle. alpha Myh6 1.7 down 0.008 1427168_a_at procollagen. type XIV. alpha 1 Col14a1 1.7 down 0.005 1423607_at lumican Lum 1.7 down 0.005 1451310_a_at cathepsin L Ctsl 1.7 down 0.001 1452363_a_at ATPase.