UC Irvine UC Irvine Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acidity, Basicity, and Pka 8 Connections
Acidity, basicity, and pKa 8 Connections Building on: Arriving at: Looking forward to: • Conjugation and molecular stability • Why some molecules are acidic and • Acid and base catalysis in carbonyl ch7 others basic reactions ch12 & ch14 • Curly arrows represent delocalization • Why some acids are strong and others • The role of catalysts in organic and mechanisms ch5 weak mechanisms ch13 • How orbitals overlap to form • Why some bases are strong and others • Making reactions selective using conjugated systems ch4 weak acids and bases ch24 • Estimating acidity and basicity using pH and pKa • Structure and equilibria in proton- transfer reactions • Which protons in more complex molecules are more acidic • Which lone pairs in more complex molecules are more basic • Quantitative acid/base ideas affecting reactions and solubility • Effects of quantitative acid/base ideas on medicine design Note from the authors to all readers This chapter contains physical data and mathematical material that some readers may find daunting. Organic chemistry students come from many different backgrounds since organic chemistry occu- pies a middle ground between the physical and the biological sciences. We hope that those from a more physical background will enjoy the material as it is. If you are one of those, you should work your way through the entire chapter. If you come from a more biological background, especially if you have done little maths at school, you may lose the essence of the chapter in a struggle to under- stand the equations. We have therefore picked out the more mathematical parts in boxes and you should abandon these parts if you find them too alien. -

Organic Functional Group Analysis
Experiment 1 1 Laboratory Experiments for GOB Chemistry ___________________________________________________________________________________________ I ORGANIC FUNCTIONAL GROUP ANALYSIS I. OBJECTIVES AND BACKGROUND This experiment will introduce you to some of the more common functional groups of organic chemistry. The functional group is that portion of the molecule that undergoes a structural change during a chemical reaction. The functional groups that will be studied in this experiment are carboxylic acid, amines aldehyde, ketone, alcohols and alkenes. You will learn chemical tests that will allow you to distinguish one functional group from another. You will use the chemical tests to identify the functionality of an unknown organic compound. In addition, you will use a water solubility test to determine whether your organic compound is of high or low formula weight. The chemical tests you will perform make up a sequence of experiments designed to determine the absence of or suggest the presence of particular functional groups. The complete sequence is shown in the flow diagram on page 8. This diagram can serve you in several ways: It is a summary of the procedure that you are to follow in classifying your unknown as one of the functional group types. It can order your thoughts as you read the discussion of each test, and help you to understand the significance of that test. It can enhance your appreciation for and enjoyment of this experiment. Your role is that of chemist and detective: you will employ this cleverly devised scheme to sleuth out the identity of your unknown's functionality. Experiment 1 2 Laboratory Experiments for GOB Chemistry ___________________________________________________________________________________________ Discussion of Chemical Tests 1. -

Comparative Strengths of Four Organic Bases in Benzene1 Marion Maclean Davis and Hannah B
Journal of Research of the National Bureau of Standards Vol. 48, No. 5, May 1952 Research Paper 2326 Comparative Strengths of Four Organic Bases in Benzene1 Marion Maclean Davis and Hannah B. Hetzer Spectropho to metric studies have shown that the reaction of the base 1,3-di-o-tolylguani- dine with the acidic indicator dye bromophthalein magenta E (tetrabromophenolphthalein ethyl ester) in benzene at 25° C, like the reactions of 1,3-diphenylguanidine and 1,2,3-tri- phenylguanidine with the same indicator, can be represented by the following two equations: B + HA ^ BH+.A- (colorless base) (yellow acid) (magenta salt) BH+.A- + B^(BHB)+A- (blue salt) For ditolylguanidine, the equilibrium constants K\ and K2 for the first and second reactions, respectively, are estimated to be 1.1 X106 and 6.4. These values are compared with values for Ki and K2 previously found for di- and triphenylguanidine and the value of Ki found for triethylamine. The values for K\, which measure the relative tendencies of the bases to form salts with the indicator acid in benzene, would be expected to parallel the ionic dissociation constants of the bases in water. However, the parallelism is not good. Diphenylguanidine and ditolylguariidine, which are presumed to be weaker bases in water than triethylamine, are much more reactive in benzene. The results demonstrate how misleading the aqueous dissociation constants may be as a gage of the relative reactivities of bases in a nonaqueous solvent such as benzene. Steric and solvation effects are discussed. 1. Introduction i optical instruments then available, it was possible to make only roughly quantitative comparisons of Hantzsch and his coworkers were the first to J acidic strengths. -

Chapter 14 – Acid-Base Equilibria Most Solutions That Occur in Nature Are Slightly Acidic. One Reason for This Is That When C
Chapter 14 – Acid-Base Equilibria Most solutions that occur in nature are slightly acidic. One reason for this is that when carbon dioxide dissolves in water, it forms carbonic acid, H2CO3. Basic solutions that exist in nature typically have limited direct exposure to air. For example, your blood is slightly basic and, under ideal circumstances, is not open to the air. Likewise, peach pits contain strychnine, an organic base, which is not protonated because of the shell that seals out the atmosphere. Figure 14.2 (p. 769) lists the pHs of some common substances. Note that only seawater occurs openly in nature with a pH over 7. We’ll see why before this chapter is through. 14.1 Brønsted-Lowry Acids and Bases Earlier, in Chapter 2, you were given quick working definitions of acids and bases. We will now expand on those definitions, but begin with a short reminder of how acids and bases were originally identified and characterized. Acidic solutions generally have a sour taste (e.g. grapefruit and lemon juices) and dissolve certain metals. Bases have a bitter taste and feel slippery. The slippery feeling comes from the base breaking down oils and fatty acids in your hands. The old lye soap used a century ago cleaned by removing the top layer of skin and anything that was attached to it. Modern soaps use a different type of active chemical that doesn’t react with your skin. The first good definition of acids and bases was proposed by Arrhenius (the same person who proposed activation energy). He proposed that acids were substances that produces H+ ions in water and bases were substances that produces OH- ions in water. -

Types of Titrations 1- Acid–Base Titration
Types of titrations There are many types of titrations with different procedures and goals. The most common types of qualitative titration are acid–base titrations and redox titrations. 1- Acid–base titration Neutralization titrations are widely used to determine the amounts of acids and bases and to monitor the progress of reactions that produce or consume hydrogen ions. In addition, we investigate titration curves that are plots of pH vs. volume of titrant, and present several examples of pH calculations. Acid–base titrations depend on the neutralization between an acid and a base when mixed in solution. In addition to the sample, an appropriate pH indicator is added to the titration chamber, reflecting the pH range of the equivalence point. The acid–base indicator indicates the endpoint of the titration by changing color. The endpoint and the equivalence point are not exactly the same because the equivalence point is determined by the stoichiometry of the reaction while the endpoint is just the color change from the indicator. Thus, a careful selection of the indicator will reduce the indicator error. For example, if the equivalence point is at a pH of 8.4, then the Phenolphthalein indicator would be used instead of Alizarin Yellow because phenolphthalein would reduce the indicator error. Common indicators, their colors, and the pH range in which they change color are given in the table above.[23] When more precise results are required, or when the reagents are a weak acid and a weak base, a pH meter or a conductance meter are used. For very strong bases, such as organolithium reagent, metal amides, and hydrides, water is generally not a suitable solvent and indicators whose pKa are in the range of aqueous pH changes are of little use. -

Shifted Equilibria of Organic Acids and Bases in the Aqueous Surface Region† Cite This: Phys
PCCP View Article Online PAPER View Journal | View Issue Shifted equilibria of organic acids and bases in the aqueous surface region† Cite this: Phys. Chem. Chem. Phys., 2018, 20,23281 ab b a Josephina Werner, * Ingmar Persson, Olle Bjo¨rneholm, Delphine Kawecki,‡a Clara-Magdalena Saak, a Marie-Madeleine Walz, §a Victor Ekholm, a Isaak Unger,a Corina Valtl,a Carl Caleman, ac Gunnar O¨ hrwall d and Nønne L. Prisle ef Acid–base equilibria of carboxylic acids and alkyl amines in the aqueous surface region were studied using surface-sensitive X-ray photoelectron spectroscopy and molecular dynamics simulations. Solutions of these organic compounds were examined as a function of pH, concentration and chain length to investigate the distribution of acid and base form in the surface region as compared to the aqueous bulk. Results from these experiments show that the neutral forms of the studied acid–base pairs are strongly enriched in the aqueous surface region. Moreover, we show that for species with at Creative Commons Attribution-NonCommercial 3.0 Unported Licence. least four carbon atoms in their alkyl-chain, their charged forms are also found to be abundant in the surface region. Using a combination of XPS and MD results, a model is proposed that effectively describes the surface composition. Resulting absolute surface concentration estimations show clearly that the total organic mole fractions in the surface region change drastically as a function of solution Received 23rd March 2018, pH. The origin of the observed surface phenomena, hydronium/hydroxide concentrations in the Accepted 30th August 2018 aqueous surface region and why standard chemical equations, used to describe equilibria in dilute bulk DOI: 10.1039/c8cp01898g solution are not valid in the aqueous surface region, are discussed in detail. -

Chemical Storage Guidelines
Chemical Storage Guidelines Use these guidelines to determine appropriate storage locations for the chemicals in your area. The tables below show examples of chemicals within each group, but are NOT all inclusive. For more information about storing chemicals, refer to your Safety Data Sheets, the Laboratory Safety Manual, or contact an EH&S Laboratory Safety Specialist. Acids (pH < 7.0) Mineral acid Organic acid Oxidizing acid Hydrochloric acid Acetic acid Nitric acid Phosphoric acid Formic acid Perchloric acid Storage: Store in a corrosives cabinet, if available, or in compatible secondary containment. Incompatibility information: Acids should be segregated from bases and flammables. Oxidizing acids are incompatible with most chemicals, especially organics. Specific combinations to avoid: • Acetic acid with chromic acid, nitric acid, hydroxyl compounds, ethylene glycol, perchloric acid, peroxides, or permanganates • Chromic acid with acetic acid, naphthalene, camphor, glycerin, turpentine, alcohol, (especially ethanol) or flammable liquids • Nitric acid with acetic acid, aniline, chromic acid, hydrocyanic acid, hydrogen sulfide, flammable liquids, flammable gases, copper, brass, or any heavy metals • Perchloric acid with acetic acid, acetic anhydride, bismuth and its alloys, alcohol, paper, wood, ether, oils or grease Bases (pH > 7.0) Inorganic base Organic base Potassium hydroxide Diethylamine Sodium hydroxide Piperidine Storage: Store in a corrosives cabinet, if available, or in compatible secondary containment. Incompatibility information: Bases should be segregated from acids, flammables, and reactives. Environmental Health and Safety | 2408 Wanda Daley Drive | Ames, IA 50011-3602 | Ph: (515) 294-5359 | www.ehs.iastate.edu Reviewed 2018 1 Flammables Flammable liquid Flammable solid Acetone Napthalene Ether Paraformaldehyde Storage: Flammable liquids totaling more than 10 gallons must be stored in a flammable cabinet. -

Acid/Base Indicators
Chapter 14 Principles of Neutralization Titrations Neutralization titrations are widely used to determine the amounts of acids and bases and to monitor the progress of reactions that produce or consume hydrogen ions. This chapter explores the principles of acid/base titrations. In addition, we investigate titration curves that are plots of pH vs. volume of titrant, and present several examples of pH calculations. 14 A Solutions and indicators for acid/base titrations Neutralization titrations depend on a chemical reaction of the analyte with a standard reagent. There are several different types of acid/base titrations. 1. The titration of a strong acid, such as hydrochloric or sulfuric acid, with a strong base, such as sodium hydroxide. 2. The titration of a weak acid, such as acetic or lactic acid, with a strong base. 3. The titration of a weak base, such as sodium cyanide or sodium salicylate, with a strong acid. • In all titrations, we must have a method of determining the point of chemical equivalence • Typically, a chemical indicator or an instrumental method is used to locate the end point, which we hope is very close to the equivalence point. Our discussion focuses on the types of standard solutions and the chemical indicators that are used for neutralization titr. ns Standard Solutions The standard reagents used in acid/base titrations are always strong acids or strong bases, such as HCl, HClO4, H2SO4, NaOH, and KOH. Weak acids and bases are never used as standard reagents because they react incompletely with analytes. Standard solutions of acids are prepared by diluting concentrated hydrochloric, perchloric, or sulfuric acid. -

Download Article (PDF)
Utilization of the Sterically Hindered Base, 4-Hydroxy-2,2,6,6-tetramethylpiperidine, as a Hydrogen Halide Acceptor George Sosnovsky and Maria Konieczny* Department of Chemistry, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53201, USA Z. Naturforsch. 33 b, 792-796 (1978); received March 17, 1978 Dehydrobromination, Preparation of Alkenes, Quaternization of Amines, Sterically Hindered Bases, 4-Hydroxy-2,2,6,6-tetramethylpiperidine The synthetic utility of the sterically hindered base, 4-hydroxy-2,2,6,6-tetramethyl- piperidine (1) as a hydrogen halide acceptor was studied. HINSKT"^^ 3 3 H3C | CH3 ' RN(CH ) F H 1 2 It was shown that base 1 can be effectively used in the quaternization reaction of primary aliphatic and aromatic amines to give the corresponding trimethylammonium iodides 2 in 88-98% yield. Base 1 was used also as the dehydrobrominating agent of alkyl and cyclo- alkyl bromides to give the corresponding unsaturated compounds in 82-95% yield. A. Exhaustive Methylation 0! Amines propylaniline [6], and 1,2,2,6,6-pentamethylpiper- idine (PMP) [1, 2, 6] have been utilized in the Over the years, methods have been developed exhaustive alkylation reactions of primary amines. for the preparation of quaternary ammonium com- The organic bases are superior to inorganic bases, pounds, which are important as organic inter- such as, sodium hydroxide due to the ease of sep- mediates, detergents, insecticides, bacteriostats, aration of by-products and lack of complicated side and drugs [1, 2]. The most common method for reactions under the milder reaction conditions. preparation of these compounds is by alkylation Sommer et al. -

Analytical Chemistry
Analytical chemistry Denise Lowinsohn [email protected] http://www.ufjf.br/nupis 2018 SCHEDULE Date Activities 21/03/18 -Aqueous-solution chemistry 28/03/18 -Neutralization tritations 04/04/18 -Precipitation 11/04/18 -Precipitation tritimetry 18/04/18 -Oxidation/reduction 25/04/18 -Oxidation/reduction titration 02/05/18 -Complex-formation titrations: part 1 -Complex-formation titrations: part 2 09/05/18 -Preparing samples for analysis 16/05/18 1º Test (100 points) 23/05/18 -Preparing samples for analysis -Preparing samples for analysis: activity delivery (100 points) 30/05/18 -Application of Statistics to Analytical Chemistry 06/06/18 -Application of Statistics to Analytical Chemistry 13/06/18 Holiday -Application of Statistics to Analytical Chemistry 20/06/18 -Sampling experiment (100 points) 27/06/18 2º Test (100 points) 04/07/18 Oral presentation (100 points) Aqueous-solution chemistry References Brown, LeMay e Bursten, Química - A ciência central, 9ª edição, Editora Pearson – Prentice Hall, 2005. Daniel C. Harris, Análise Química Quantitativa, Editora LTC, 5a edição, 2001. Skoog, West, Holler, Fundamentals of analytical chemistry, 7th edition, 1995. Acid-base equilibrium The behaviour of acids and bases is very important in all areas of Chemistry and others areas of Science. Industrial processes, Laboratory and Biological Effect of pH - The pH of the medium is an extremely important parameter for many reactions in Analytical Chemistry. Acid and base: a brief review Acid: taste sour and cause colour changes in pigments. Base: bitter taste and slippery feeling. Arrhenius: In aqueous medium, acids are defined as substances that increase [H+] and bases increase [OH-] + + Acids = substances that produce H3O (H ) ions, when dissolved in water Bases = substances that produce OH- ions, when dissolved in water Arrhenius: acid + base salt + water. -
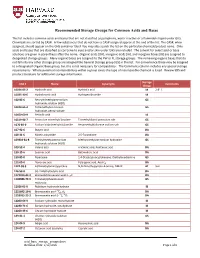
Common Acid and Base Storage Group Recommendations
Recommended Storage Groups for Common Acids and Bases This list includes common acids and bases that are not classified as pyrophoric, water reactive or a flammable liquid under GHS. Chemicals are sorted by CAS#. A few substances that do not have a CAS# assigned appear at the end of the list. The CAS#, when assigned, should appear on the GHS container label. You may also search the list on the particular chemical/product name. Only acids and bases that are classified as corrosive to eyes and/or skin under GHS are included. The solvent for select acid or base solutions are given in parenthesis after the name. Organic acids [OA], inorganic acids [IA], and inorganic bases [IB] are assigned to designated storage groups. Many organic bases are assigned to the PW or FL storage groups. The remaining organic bases that do not fall into any other storage group are assigned the General Storage group [GS] in this list. For convenience these may be assigned to a designated Organic Base group, but this is not necessary for compatibility. The Comments column includes any special storage requirements. Where potential incompatibilities within a group exists the type of incompatible chemical is listed. Review SDS and product literature for additional storage information. Storage CAS # Name Synonyms Comments Group 10034-85-2 Hydriodic acid Hydriotic acid IA 2-8° C 10035-10-6 Hydrobromic acid Hydrogen Bromide IA 100-85-6 Benzyltrimethylammonium GS hydroxide solution (H2O) 10424-65-4 Tetramethylammonium GS hydroxide, pentahydrate 10450-60-9 Periodic acid -

NOTES – Acids, Bases and Ph, Chapter 14
NOTES – Acids, Bases and pH, Chapter 14 Terms: Acid dissociation constant, Ka the equilibrium constant for a reaction in which a proton is removed + from an acid by the H2O to form a conjugate base and H30 Acidic oxide a covalent oxide that dissolves in water to give an acidic solution Amine: an organic base derived from ammonia in which one or more of the hydrogen atoms are replaced by organic groups Amphoteric substance a substance that can behave either as an acid or a base Basic oxide an ionic oxide that dissolves in water to produce a basic solution Carboxyl group the –COOH group in an organic acid Conjugate base what remains of an acid molecule after a proton is lost Conjugate acid the species formed when a proton is added to a base Conjugate acid-base pair two species related to each other by the donating and accepting of a single proton Diprotic acid an acid having two acidic protons (ex: sulfuric acid) + Hydronium Ion the H30 ion; a hydrated proton Ion- product constant, (Kw) The equilibrium constant for the auto ionization of water; (Kw)= + - -14 [H ][OH ] at 25 ºC. (Kw)= 1.0x10 Lewis Acid an electron-pair acceptor Lewis base an electron-pair donor Lime-soda process a water-softening method in which lime and soda ash are added to water to remove calcium and magnesium ions by precipitation Monoprotic acid an acid with one acidic proton Oxyacids an acid in which the acidic proton is attached to an oxygen atom Organic acid an acid with a carbon-atom backbone; often contains the carboxyl group pH scale a log scale based on 10 and equal to –log[H+]; a convenient way to represent solution acidity that ranges from 0 - 7(neutral/H2O) - 14 Major species the components present in relatively large amounts in a solution Percent dissociation the ratio of the amount of a substance that is dissociated at equilibrium to the initial concentration of the substance in the solution multiplied by 100.