Synthesis of Phylogeny and Taxonomy Into a Comprehensive Tree of Life
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Foucault's Darwinian Genealogy
genealogy Article Foucault’s Darwinian Genealogy Marco Solinas Political Philosophy, University of Florence and Deutsches Institut Florenz, Via dei Pecori 1, 50123 Florence, Italy; [email protected] Academic Editor: Philip Kretsedemas Received: 10 March 2017; Accepted: 16 May 2017; Published: 23 May 2017 Abstract: This paper outlines Darwin’s theory of descent with modification in order to show that it is genealogical in a narrow sense, and that from this point of view, it can be understood as one of the basic models and sources—also indirectly via Nietzsche—of Foucault’s conception of genealogy. Therefore, this essay aims to overcome the impression of a strong opposition to Darwin that arises from Foucault’s critique of the “evolutionistic” research of “origin”—understood as Ursprung and not as Entstehung. By highlighting Darwin’s interpretation of the principles of extinction, divergence of character, and of the many complex contingencies and slight modifications in the becoming of species, this essay shows how his genealogical framework demonstrates an affinity, even if only partially, with Foucault’s genealogy. Keywords: Darwin; Foucault; genealogy; natural genealogies; teleology; evolution; extinction; origin; Entstehung; rudimentary organs “Our classifications will come to be, as far as they can be so made, genealogies; and will then truly give what may be called the plan of creation. The rules for classifying will no doubt become simpler when we have a definite object in view. We possess no pedigrees or armorial bearings; and we have to discover and trace the many diverging lines of descent in our natural genealogies, by characters of any kind which have long been inherited. -

Principles of Plant Taxonomy Bot
PRINCIPLES OF PLANT TAXONOMY BOT 222 Dr. M. Ajmal Ali, PhD 1 What is Taxonomy / Systematics ? Animal group No. of species Amphibians 6,199 Birds 9,956 Fish 30,000 Mammals 5,416 Tundra Reptiles 8,240 Subtotal 59,811 Grassland Forest Insects 950,000 Molluscs 81,000 Q: Why we keep the stuffs of our home Crustaceans 40,000 at the fixed place or arrange into some Corals 2,175 kinds of system? Desert Others 130,200 Rain forest Total 1,203,375 • Every Human being is a Taxonomist Plants No. of species Mosses 15,000 Ferns and allies 13,025 Gymnosperms 980 Dicotyledons 199,350 Monocotyledons 59,300 Green Algae 3,715 Red Algae 5,956 Lichens 10,000 Mushrooms 16,000 Brown Algae 2,849 Subtotal 28,849 Total 1,589,361 • We have millions of different kind of plants, animals and microorganism. We need to scientifically identify, name and classify all the living organism. • Taxonomy / Systematics is the branch of science deals with classification of organism. 2 • Q. What is Plant Taxonomy / Plant systematics We study plants because: Plants convert Carbon dioxide gas into Every things we eat comes Plants produce oxygen. We breathe sugars through the process of directly or indirectly from oxygen. We cannot live without photosynthesis. plants. oxygen. Many chemicals produced by the Study of plants science helps to Study of plants science helps plants used as learn more about the natural Plants provide fibres for paper or fabric. to conserve endangered medicine. world plants. We have millions of different kind of plants, animals and microorganism. -

Ten Misunderstandings About Evolution a Very Brief Guide for the Curious and the Confused by Dr
Ten Misunderstandings About Evolution A Very Brief Guide for the Curious and the Confused By Dr. Mike Webster, Dept. of Neurobiology and Behavior, Cornell Lab of Ornithology, Cornell University ([email protected]); February 2010 The current debate over evolution and “intelligent design” (ID) is being driven by a relatively small group of individuals who object to the theory of evolution for religious reasons. The debate is fueled, though, by misunderstandings on the part of the American public about what evolutionary biology is and what it says. These misunderstandings are exploited by proponents of ID, intentionally or not, and are often echoed in the media. In this booklet I briefly outline and explain 10 of the most common (and serious) misunderstandings. It is impossible to treat each point thoroughly in this limited space; I encourage you to read further on these topics and also by visiting the websites given on the resource sheet. In addition, I am happy to send a somewhat expanded version of this booklet to anybody who is interested – just send me an email to ask for one! What are the misunderstandings? 1. Evolution is progressive improvement of species Evolution, particularly human evolution, is often pictured in textbooks as a string of organisms marching in single file from “simple” organisms (usually a single celled organism or a monkey) on one side of the page and advancing to “complex” organisms on the opposite side of the page (almost invariably a human being). We have all seen this enduring image and likely have some version of it burned into our brains. -

The Taxonomy of Human Evolved Psychological Adaptations
Evo Edu Outreach (2012) 5:312–320 DOI 10.1007/s12052-012-0428-8 EVOLUTION AND EDUCATION RESOURCES PsychTable.org: The Taxonomy of Human Evolved Psychological Adaptations Niruban Balachandran & Daniel J. Glass Published online: 8 July 2012 # Springer Science+Business Media, LLC 2012 Abstract We announce the launch of PsychTable.org,a Introduction collaborative web-based project devoted to classifying and evaluating evolved psychological adaptations The timing is right to establish a classification system for (EPAs), geared toward researchers, educators, students, human evolved psychological adaptations (EPAs). Several and the general public. The website works by aggregat- factors have coalesced to create a unique opportunity to de- ing citations which support or challenge the existence of velop a taxonomy that will facilitate the expansion of a culture each purported EPA, using a mathematical algorithm to of both synthesis and empirical rigor in the evolutionary assign an evidentiary strength score to each, and gener- behavioral sciences, including disciplines such as evolution- ating a table which represents the current but ever- ary cognitive neuroscience, behavioral ecology, “evo-devo,” changing state of the empirical evidence. Citations are primatology, evolutionary anthropology, human ethology, ge- added and assigned evaluative ratings by both general netics, and others. users and an international community of expert contrib- These aforementioned factors include: a large international utors; as such, the content of the site will represent the body of research describing hundreds of amassed EPAs; a consensus of the scientific community and new research global research community of evolutionary behavioral scien- opportunities. PsychTable has features for achieving empir- tists that recognizes the importance of both synthesis and ical meta-goals such as quality control, hypothesis testing, classification; the ascent of evolutionary psychology as a cross-disciplinary collaboration, and didactic utility. -

Darwin's “Tree of Life”
Icons of Evolution? Why Much of What Jonathan Wells Writes about Evolution is Wrong Alan D. Gishlick, National Center for Science Education DARWIN’S “TREE OF LIFE” mon descent. Finally, he demands that text- books treat universal common ancestry as PHYLOGENETIC TREES unproven and refrain from illustrating that n biology, a phylogenetic tree, or phyloge- “theory” with misleading phylogenies. ny, is used to show the genealogic relation- Therefore, according to Wells, textbooks Iships of living things. A phylogeny is not should state that there is no evidence for com- so much evidence for evolution as much as it mon descent and that the most recent research is a codification of data about evolutionary his- refutes the concept entirely. Wells is complete- tory. According to biological evolution, organ- ly wrong on all counts, and his argument is isms share common ancestors; a phylogeny entirely based on misdirection and confusion. shows how organisms are related. The tree of He mixes up these various topics in order to life shows the path evolution took to get to the confuse the reader into thinking that when current diversity of life. It also shows that we combined, they show an endemic failure of can ascertain the genealogy of disparate living evolutionary theory. In effect, Wells plays the organisms. This is evidence for evolution only equivalent of an intellectual shell game, put- in that we can construct such trees at all. If ting so many topics into play that the “ball” of evolution had not happened or common ances- evolution gets lost. try were false, we would not be able to discov- THE CAMBRIAN EXPLOSION er hierarchical branching genealogies for ells claims that the Cambrian organisms (although textbooks do not general- Explosion “presents a serious chal- ly explain this well). -

The Rooting of the Universal Tree of Life Is Not Reliable
J Mol Evol (1999) 49:509–523 © Springer-Verlag New York Inc. 1999 The Rooting of the Universal Tree of Life Is Not Reliable Herve´ Philippe,1 Patrick Forterre2 1 Phyloge´nie et Evolution Mole´culaires (UPRESA 8080 CNRS), Baˆtiment 444, Universite´ Paris-Sud, 91405 Orsay-Cedex, France 2 Institut de Ge´ne´tique et Microbiologie (UMR 8621 CNRS), Baˆtiment 409, Universite´ Paris-Sud, 91405 Orsay-Cedex, France Abstract. Several composite universal trees connected terial rooting could be explained by an attraction be- by an ancestral gene duplication have been used to root tween this branch and the long branch of the outgroup. the universal tree of life. In all cases, this root turned out Finally, we suggested that an eukaryotic rooting could be to be in the eubacterial branch. However, the validity of a more fruitful working hypothesis, as it provides, for results obtained from comparative sequence analysis has example, a simple explanation to the high genetic simi- recently been questioned, in particular, in the case of larity of Archaebacteria and Eubacteria inferred from ancient phylogenies. For example, it has been shown that complete genome analysis. several eukaryotic groups are misplaced in ribosomal RNA or elongation factor trees because of unequal rates Key words: Root of the tree of life — ATPase — of evolution and mutational saturation. Furthermore, the Carbamoyl phosphate synthetase — Elongation factor — addition of new sequences to data sets has often turned tRNA synthetase — Signal recognition particle — Mu- apparently reasonable phylogenies into confused ones. tational saturation — Long branch attraction We have thus revisited all composite protein trees that have been used to root the universal tree of life up to now (elongation factors, ATPases, tRNA synthetases, car- bamoyl phosphate synthetases, signal recognition par- Introduction ticle proteins) with updated data sets. -

R E S E a R C H / M a N a G E M E N T Aquatic and Terrestrial Flatworm (Platyhelminthes, Turbellaria) and Ribbon Worm (Nemertea)
RESEARCH/MANAGEMENT FINDINGSFINDINGS “Put a piece of raw meat into a small stream or spring and after a few hours you may find it covered with hundreds of black worms... When not attracted into the open by food, they live inconspicuously under stones and on vegetation.” – BUCHSBAUM, et al. 1987 Aquatic and Terrestrial Flatworm (Platyhelminthes, Turbellaria) and Ribbon Worm (Nemertea) Records from Wisconsin Dreux J. Watermolen D WATERMOLEN Bureau of Integrated Science Services INTRODUCTION The phylum Platyhelminthes encompasses three distinct Nemerteans resemble turbellarians and possess many groups of flatworms: the entirely parasitic tapeworms flatworm features1. About 900 (mostly marine) species (Cestoidea) and flukes (Trematoda) and the free-living and comprise this phylum, which is represented in North commensal turbellarians (Turbellaria). Aquatic turbellari- American freshwaters by three species of benthic, preda- ans occur commonly in freshwater habitats, often in tory worms measuring 10-40 mm in length (Kolasa 2001). exceedingly large numbers and rather high densities. Their These ribbon worms occur in both lakes and streams. ecology and systematics, however, have been less studied Although flatworms show up commonly in invertebrate than those of many other common aquatic invertebrates samples, few biologists have studied the Wisconsin fauna. (Kolasa 2001). Terrestrial turbellarians inhabit soil and Published records for turbellarians and ribbon worms in leaf litter and can be found resting under stones, logs, and the state remain limited, with most being recorded under refuse. Like their freshwater relatives, terrestrial species generic rubric such as “flatworms,” “planarians,” or “other suffer from a lack of scientific attention. worms.” Surprisingly few Wisconsin specimens can be Most texts divide turbellarians into microturbellarians found in museum collections and a specialist has yet to (those generally < 1 mm in length) and macroturbellari- examine those that are available. -

Sociobiology 65(4): 645-653 (October, 2018) Special Issue DOI: 10.13102/Sociobiology.V65i4.3464
Sociobiology 65(4): 645-653 (October, 2018) Special Issue DOI: 10.13102/sociobiology.v65i4.3464 Sociobiology An international journal on social insects Research article - Bees Two Colors, One Species: The Case of Melissodes nigroaenea (Apidae: Eucerini), an Important Pollinator of Cotton Fields in Brazil C Grando1,2, ND Amon2,6, SJ Clough3, N Guo3, W Wei3, P Azevedo1,4, MM López-Uribe2, MI Zucchi5 1 - Institute of Biology, University of Campinas, Campinas, São Paulo, Brazil 2 - Department of Entomology, Center for Pollinator Research, Pennsylvania State University, University Park, USA 3 - Department of Crop Science, University of Illinois at Urbana-Champaign, Urbana, USA 4 - Centro de Estudos em Insetos Sociais, Universidade Estadual Paulista “Julio de Mesquita Filho”, Rio Claro, São Paulo, Brazil 5 - Agência Paulista de Tecnologia dos Agronegócios, Pólo Regional Centro-Sul, Piracicaba, São Paulo, Brazil 6 - Department of Entomology, University of Wisconsin-Madison, Madison, USA Article History Abstract Accurate taxonomic delimitation in ecological research is absolutely critical as Edited by studies that seek to evaluate levels of biodiversity and quantify human effects on Eduardo Almeida, USP, Brazil Received 10 May 2018 the environment are rapidly undertaken. Coloration is a widely used morphological Initial acceptance 02 July 2018 character for species identification through dichotomous keys. However, taxonomic Final acceptance 26 August 2018 identification based upon coloration is often unreliable because this character can Publication date 11 October 2018 exhibit high degree of intraspecific variation. An uncertain interpretation of mesosoma color-morphs (yellow or black) occurred when we used this character, in association Keywords Melissodes Cytochrome oxidase I, Bayesian with the bristle bands in the TII, for the identification of eucerine bee phylogeny, bees, barcoding gap. -

Botanical Classification and Nomenclature an Introduction —
Botanical classification and nomenclature an introduction — Marc S.M. Sosef Jérôme Degreef Henry Engledow Pierre Meerts Botanical classification and nomenclature an introduction — Marc S.M. Sosef Jérôme Degreef Henry Engledow Pierre Meerts by Marc S.M. Sosef1, Jérôme Degreef1,2, Henry Engledow1 & Pierre Meerts3 1 Meise Botanic Garden, Nieuwelaan 38, B-1860 Meise, Belgium 2 Service Général de l’Enseignement supérieur et de la Recherche scientifique, Fédération Wallonie-Bruxelles, Rue A. Lavallée 1, B-1080 Brussels, Belgium 3 Herbarium et bibliothèque de botanique africaine, Université Libre de Bruxelles, Av. F.D. Roosevelt 50, CP 265, B-1050 Brussels, Belgium Copyright © 2020 Meise Botanic Garden, Nieuwelaan 38, 1860 Meise, Belgium. Printed in Belgium by Gewadrupo, Arendonk. This publication is published and distributed in Open Access under the Creative Commons Attribution 4.0 International license (CC-BY 4.0), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited. A PDF file of this publication can be ordered, free of charge (send an email to [email protected]), or downloaded from the webshop of Meise Botanic Garden at http://shopbotanicgarden.weezbe.com. DOI: 10.5281/zenodo.3706707 CIP Royal Library Albert I, Brussels Botanical classification and nomenclature, an introduction. Marc S.M. Sosef, Jérôme Degreef, Henry Engledow & Pierre Meerts - Meise, Meise Botanic Garden, 2020. - 72 p.; ill.; 22 x 15 cm. ISBN 9789492663207 Subject: Botany D/2020/0325/002 Content Introduction . 5 1. The history of classification . 9 1.1 Theophrastus to the Middle Ages . 11 1.2 Renaissance, Pre-Linnean period . 13 1.3 Linnaeus and the Linnaeans . -

Evidence for Evolution
CHAPTER 3 Evidence for Evolution VOLUTIONARY BIOLOGY HAS PROFOUNDLY altered our view of nature and of ourselves. At the beginning of this book, we showed the practical application of Eevolutionary biology to agriculture, biotechnology, and medicine. More broadly, evolutionary theory underpins all our knowledge of biology, explains how organisms came to be (both describing their history and identi- fying the processes that acted), and explains why they are as they are (why organisms reproduce sexually, why they age, and so on). How- ever, arguably its most important influence has been on how we view ourselves and our place in the world. The radical scope of evolution- ary biology has for many been hard to accept, and this has led to much misunderstanding and many objections. In this chapter, we summarize the evidence for evolution, clarify some common misun- derstandings, and discuss the wider implications of evolution by natural selection. Biological evolution was widely accepted soon after the publication of On the Origin of Species in 1859 (Chapter 1.x). Charles Darwin set out “one long argument” for the “descent with modification” of all liv- ing organisms, from one or a few common ancestors. He marshaled evidence from classification of organisms, from the fossil record, from geographic distribution of organisms, and by analogy with artificial se- lection. As we saw in Chapter 1, the detailed processes that cause evo- lution remained obscure until after the laws of heredity were established in the early 20th century. By the time of the Evolutionary Synthesis,in the mid-20th century, these processes were well understood and, cru- cially, it was established that adaptation is due to natural selection (Chapter 1.x). -

Definitions in Phylogenetic Taxonomy
Syst. Biol. 48(2):329–351, 1999 Denitions in PhylogeneticTaxonomy:Critique and Rationale PAUL C. SERENO Department of Organismal Biologyand Anatomy, Universityof Chicago, 1027E. 57thStreet, Chicago, Illinois 60637, USA; E-mail: [email protected] Abstract.—Ageneralrationale forthe formulation andplacement of taxonomic denitions in phy- logenetic taxonomyis proposed, andcommonly used terms such as“crown taxon”or “ node-based denition” are more precisely dened. In the formulation of phylogenetic denitions, nested refer- ence taxastabilize taxonomic content. Adenitional conguration termed a node-stem triplet also stabilizes the relationship between the trio of taxaat abranchpoint,in the face of local changein phylogenetic relationships oraddition/ deletion of taxa.Crown-total taxonomiesuse survivorship asa criterion forplacement of node-stem triplets within ataxonomic hierarchy.Diversity,morphol- ogy,andtradition alsoconstitute heuristic criteria forplacement of node-stem triplets. [Content; crown; denition; node;phylogeny; stability; stem; taxonomy.] Doesone type ofphylogenetic denition Mostphylogenetic denitions have been (apomorphy,node,stem) stabilize the taxo- constructedin the systematicliterature since nomiccontent of ataxonmore than another then withoutexplanation or justi cation for in the face oflocal change of relationships? the particulartype ofde nition used. The Isone type of phylogenetic denition more justication given forpreferential use of suitablefor clades with unresolved basalre- node- andstem-based de nitions for crown -
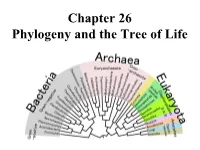
Chapter 26 Phylogeny and the Tree of Life • Biologists Estimate That There Are About 5 to 100 Million Species of Organisms Living on Earth Today
Chapter 26 Phylogeny and the Tree of Life • Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. • Evidence from morphological, biochemical, and gene sequence data suggests that all organisms on Earth are genetically related, and the genealogical relationships of living things can be represented by a vast evolutionary tree, the Tree of Life. • The Tree of Life then represents the phylogeny of organisms, the history of organismal lineages as they change through time. – In other words, phylogeny is the evolutionary history of a species or group of related species. • Phylogeny assumes that all life arise from a previous ancestors and that all organisms (bacteria, fungi, protist, plants, animals) are connected by the passage of genes along the branches of the phylogenetic tree. • The discipline of systematics classifies organisms and determines their evolutionary relationships. • Systematists use fossil, molecular, and genetic data to infer evolutionary relationships. • Hence, systematists depict evolutionary relationships among organisms as branching phylogenetic trees. • A phylogenetic tree represents a hypothesis about evolutionary relationships. • Taxonomy is the science of organizing, classifying and naming organisms. • Carolus Linnaeus was the scientist who came up with the two-part naming system (binomial system). – The first part of the name is the genus – The second part, called the specific epithet, is the species within the genus. – The first letter of the genus is capitalized, and the entire species name is italicized. • Homo sapiens or H. sapiens • All life are organize into the following taxonomic groups: domain, kingdom, phylum, class, order, family, genus, and species. (Darn Kids Playing Chess On Freeway Gets Squished).