The Rooting of the Universal Tree of Life Is Not Reliable
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Foucault's Darwinian Genealogy
genealogy Article Foucault’s Darwinian Genealogy Marco Solinas Political Philosophy, University of Florence and Deutsches Institut Florenz, Via dei Pecori 1, 50123 Florence, Italy; [email protected] Academic Editor: Philip Kretsedemas Received: 10 March 2017; Accepted: 16 May 2017; Published: 23 May 2017 Abstract: This paper outlines Darwin’s theory of descent with modification in order to show that it is genealogical in a narrow sense, and that from this point of view, it can be understood as one of the basic models and sources—also indirectly via Nietzsche—of Foucault’s conception of genealogy. Therefore, this essay aims to overcome the impression of a strong opposition to Darwin that arises from Foucault’s critique of the “evolutionistic” research of “origin”—understood as Ursprung and not as Entstehung. By highlighting Darwin’s interpretation of the principles of extinction, divergence of character, and of the many complex contingencies and slight modifications in the becoming of species, this essay shows how his genealogical framework demonstrates an affinity, even if only partially, with Foucault’s genealogy. Keywords: Darwin; Foucault; genealogy; natural genealogies; teleology; evolution; extinction; origin; Entstehung; rudimentary organs “Our classifications will come to be, as far as they can be so made, genealogies; and will then truly give what may be called the plan of creation. The rules for classifying will no doubt become simpler when we have a definite object in view. We possess no pedigrees or armorial bearings; and we have to discover and trace the many diverging lines of descent in our natural genealogies, by characters of any kind which have long been inherited. -

Synthesis of Phylogeny and Taxonomy Into a Comprehensive Tree of Life
Synthesis of phylogeny and taxonomy into a comprehensive tree of life Cody E. Hinchliffa,1, Stephen A. Smitha,1,2, James F. Allmanb, J. Gordon Burleighc, Ruchi Chaudharyc, Lyndon M. Coghilld, Keith A. Crandalle, Jiabin Dengc, Bryan T. Drewf, Romina Gazisg, Karl Gudeh, David S. Hibbettg, Laura A. Katzi, H. Dail Laughinghouse IVi, Emily Jane McTavishj, Peter E. Midfordd, Christopher L. Owenc, Richard H. Reed, Jonathan A. Reesk, Douglas E. Soltisc,l, Tiffani Williamsm, and Karen A. Cranstonk,2 aEcology and Evolutionary Biology, University of Michigan, Ann Arbor, MI 48109; bInterrobang Corporation, Wake Forest, NC 27587; cDepartment of Biology, University of Florida, Gainesville, FL 32611; dField Museum of Natural History, Chicago, IL 60605; eComputational Biology Institute, George Washington University, Ashburn, VA 20147; fDepartment of Biology, University of Nebraska-Kearney, Kearney, NE 68849; gDepartment of Biology, Clark University, Worcester, MA 01610; hSchool of Journalism, Michigan State University, East Lansing, MI 48824; iBiological Science, Clark Science Center, Smith College, Northampton, MA 01063; jDepartment of Ecology and Evolutionary Biology, University of Kansas, Lawrence, KS 66045; kNational Evolutionary Synthesis Center, Duke University, Durham, NC 27705; lFlorida Museum of Natural History, University of Florida, Gainesville, FL 32611; and mComputer Science and Engineering, Texas A&M University, College Station, TX 77843 Edited by David M. Hillis, The University of Texas at Austin, Austin, TX, and approved July 28, 2015 (received for review December 3, 2014) Reconstructing the phylogenetic relationships that unite all line- published phylogenies are available only as journal figures, rather ages (the tree of life) is a grand challenge. The paucity of homologous than in electronic formats that can be integrated into databases and character data across disparately related lineages currently renders synthesis methods (7–9). -

Ten Misunderstandings About Evolution a Very Brief Guide for the Curious and the Confused by Dr
Ten Misunderstandings About Evolution A Very Brief Guide for the Curious and the Confused By Dr. Mike Webster, Dept. of Neurobiology and Behavior, Cornell Lab of Ornithology, Cornell University ([email protected]); February 2010 The current debate over evolution and “intelligent design” (ID) is being driven by a relatively small group of individuals who object to the theory of evolution for religious reasons. The debate is fueled, though, by misunderstandings on the part of the American public about what evolutionary biology is and what it says. These misunderstandings are exploited by proponents of ID, intentionally or not, and are often echoed in the media. In this booklet I briefly outline and explain 10 of the most common (and serious) misunderstandings. It is impossible to treat each point thoroughly in this limited space; I encourage you to read further on these topics and also by visiting the websites given on the resource sheet. In addition, I am happy to send a somewhat expanded version of this booklet to anybody who is interested – just send me an email to ask for one! What are the misunderstandings? 1. Evolution is progressive improvement of species Evolution, particularly human evolution, is often pictured in textbooks as a string of organisms marching in single file from “simple” organisms (usually a single celled organism or a monkey) on one side of the page and advancing to “complex” organisms on the opposite side of the page (almost invariably a human being). We have all seen this enduring image and likely have some version of it burned into our brains. -

Darwin's “Tree of Life”
Icons of Evolution? Why Much of What Jonathan Wells Writes about Evolution is Wrong Alan D. Gishlick, National Center for Science Education DARWIN’S “TREE OF LIFE” mon descent. Finally, he demands that text- books treat universal common ancestry as PHYLOGENETIC TREES unproven and refrain from illustrating that n biology, a phylogenetic tree, or phyloge- “theory” with misleading phylogenies. ny, is used to show the genealogic relation- Therefore, according to Wells, textbooks Iships of living things. A phylogeny is not should state that there is no evidence for com- so much evidence for evolution as much as it mon descent and that the most recent research is a codification of data about evolutionary his- refutes the concept entirely. Wells is complete- tory. According to biological evolution, organ- ly wrong on all counts, and his argument is isms share common ancestors; a phylogeny entirely based on misdirection and confusion. shows how organisms are related. The tree of He mixes up these various topics in order to life shows the path evolution took to get to the confuse the reader into thinking that when current diversity of life. It also shows that we combined, they show an endemic failure of can ascertain the genealogy of disparate living evolutionary theory. In effect, Wells plays the organisms. This is evidence for evolution only equivalent of an intellectual shell game, put- in that we can construct such trees at all. If ting so many topics into play that the “ball” of evolution had not happened or common ances- evolution gets lost. try were false, we would not be able to discov- THE CAMBRIAN EXPLOSION er hierarchical branching genealogies for ells claims that the Cambrian organisms (although textbooks do not general- Explosion “presents a serious chal- ly explain this well). -

Evidence for Evolution
CHAPTER 3 Evidence for Evolution VOLUTIONARY BIOLOGY HAS PROFOUNDLY altered our view of nature and of ourselves. At the beginning of this book, we showed the practical application of Eevolutionary biology to agriculture, biotechnology, and medicine. More broadly, evolutionary theory underpins all our knowledge of biology, explains how organisms came to be (both describing their history and identi- fying the processes that acted), and explains why they are as they are (why organisms reproduce sexually, why they age, and so on). How- ever, arguably its most important influence has been on how we view ourselves and our place in the world. The radical scope of evolution- ary biology has for many been hard to accept, and this has led to much misunderstanding and many objections. In this chapter, we summarize the evidence for evolution, clarify some common misun- derstandings, and discuss the wider implications of evolution by natural selection. Biological evolution was widely accepted soon after the publication of On the Origin of Species in 1859 (Chapter 1.x). Charles Darwin set out “one long argument” for the “descent with modification” of all liv- ing organisms, from one or a few common ancestors. He marshaled evidence from classification of organisms, from the fossil record, from geographic distribution of organisms, and by analogy with artificial se- lection. As we saw in Chapter 1, the detailed processes that cause evo- lution remained obscure until after the laws of heredity were established in the early 20th century. By the time of the Evolutionary Synthesis,in the mid-20th century, these processes were well understood and, cru- cially, it was established that adaptation is due to natural selection (Chapter 1.x). -
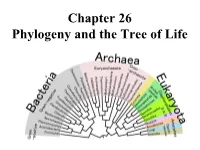
Chapter 26 Phylogeny and the Tree of Life • Biologists Estimate That There Are About 5 to 100 Million Species of Organisms Living on Earth Today
Chapter 26 Phylogeny and the Tree of Life • Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. • Evidence from morphological, biochemical, and gene sequence data suggests that all organisms on Earth are genetically related, and the genealogical relationships of living things can be represented by a vast evolutionary tree, the Tree of Life. • The Tree of Life then represents the phylogeny of organisms, the history of organismal lineages as they change through time. – In other words, phylogeny is the evolutionary history of a species or group of related species. • Phylogeny assumes that all life arise from a previous ancestors and that all organisms (bacteria, fungi, protist, plants, animals) are connected by the passage of genes along the branches of the phylogenetic tree. • The discipline of systematics classifies organisms and determines their evolutionary relationships. • Systematists use fossil, molecular, and genetic data to infer evolutionary relationships. • Hence, systematists depict evolutionary relationships among organisms as branching phylogenetic trees. • A phylogenetic tree represents a hypothesis about evolutionary relationships. • Taxonomy is the science of organizing, classifying and naming organisms. • Carolus Linnaeus was the scientist who came up with the two-part naming system (binomial system). – The first part of the name is the genus – The second part, called the specific epithet, is the species within the genus. – The first letter of the genus is capitalized, and the entire species name is italicized. • Homo sapiens or H. sapiens • All life are organize into the following taxonomic groups: domain, kingdom, phylum, class, order, family, genus, and species. (Darn Kids Playing Chess On Freeway Gets Squished). -

Answers to Common Misconceptions About Biological Evolution Leah M
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Papers in Evolution Papers in the Biological Sciences 5-2017 Answers to common misconceptions about biological evolution Leah M. Abebe University of Nebraska-Lincoln Blake Bartels University of Nebraska-Lincoln Kaitlyn M. Caron University of Nebraska-Lincoln Adam M. Gleeson University of Nebraska-Lincoln Samuel N. Johnson University of Nebraska-Lincoln See next page for additional authors Follow this and additional works at: http://digitalcommons.unl.edu/bioscievolution Part of the Evolution Commons Abebe, Leah M.; Bartels, Blake; Caron, Kaitlyn M.; Gleeson, Adam M.; Johnson, Samuel N.; Kluza, Tyler J.; Knopik, Nicholas W.; Kramer, Kristen N.; Maza, Masiel S.; Stava, Kaitlyn A.; Sullivan, Kaitlyn; Trimble, Jordan T.; and Zink, Robert M. , editor, "Answers to common misconceptions about biological evolution" (2017). Papers in Evolution. 4. http://digitalcommons.unl.edu/bioscievolution/4 This Article is brought to you for free and open access by the Papers in the Biological Sciences at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Papers in Evolution by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Leah M. Abebe; Blake Bartels; Kaitlyn M. Caron; Adam M. Gleeson; Samuel N. Johnson; Tyler J. Kluza; Nicholas W. Knopik; Kristen N. Kramer; Masiel S. Maza; Kaitlyn A. Stava; Kaitlyn Sullivan; Jordan T. Trimble; and Robert M. Zink , editor This article is available at DigitalCommons@University of Nebraska - Lincoln: http://digitalcommons.unl.edu/bioscievolution/4 Answers to common misconceptions about biological evolution Class of BIOS 472, University of Nebraska, Lincoln, Spring 2017 Students: Leah M. Abebe, Blake Bartels, Kaitlyn M. -

Chapter 1 of Philosophy and the Tree of Life
PHILOSOPHY AND THE TREE OF LIFE THE METAPHYSICS AND EPISTEMOLOGY OF PHYLOGENETIC SYSTEMATICS Joel D. Velasco PHILOSOPHY AND THE TREE OF LIFE: THE METAPHYSICS AND EPISTEMOLOGY OF PHYLOGENETIC SYSTEMATICS by Joel D. Velasco A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Philosophy) at the UNIVERSITY OF WISCONSIN-MADISON 2008 © Copyright by Joel Velasco 2008 All Rights Reserved i Abstract: This dissertation examines the foundations of phylogenetic systematics which involves both the construction of phylogenetic trees to represent evolutionary history and the use of those trees to study various aspects of that history. I begin by defending a genealogy-based view of biological taxonomy: the view that all taxa—the formal groups in our classification system—must be monophyletic, i.e., they must consist of an ancestor and all of its descendants. Furthermore, I argue that, contrary to current practice, these taxa should not be assigned ranks (such as genus, family, and order). I then proceed by applying these principles to the debate about species. I argue that non- genealogically based species concepts (such as the popular “biological species concept”) are unacceptable. Instead, a species concept must delimit species so that they form genealogically exclusive groups – groups of organisms more closely related to each other than to any organisms outside the group. With this in mind, I develop two distinct phylogenetic species concepts. Each treats a species as a genealogically exclusive group of organisms. The first determines genealogical relatedness in terms of recency of common ancestry; the second understands genealogy as a composite of gene histories. -

Phylogeny of Life
1/3/2020 TAXONOMY: RECONSTRUCTING THE TREE OF LIFE Michael L. Draney, Ph.D. Professor-Biology Chair, Department of Natural & Applied Sciences UW-Green Bay LLI May 2019 6 January 2020 Agenda/Outline ■ Introduction ■ The Tree of Life ■ Break at 11 am ■ Reconstructing and Using the TOL Tree of Life 2 1 1/3/2020 Biology: The Scientific Study of Life ■ Life: The most awe-inspiring phenomenon in the universe! – Arguably the most interesting and most beautiful phenomena in the in the universe – So far is ONLY known here on Earth ■ Almost certainly occurs elsewhere… ■ Obviously attractive to many scientists: – We call ourselves Biologists. Tree of Life 3 The most complex phenomenon in the universe ■ This complexity has resulted in astounding diversity, at different levels of organization: – Ecosystem/Habitat diversity: No two places alike – Species diversity: Each a “masterpiece” of evolution – Genetic diversity: Each species is highly variable Tree of Life 4 2 1/3/2020 Habitat/Ecosystem Diversity Tree of Life 5 Species Diversity 6 3 1/3/2020 Genetic Diversity What are the most profound and important discoveries about life biologists have made? Tree of Life 8 4 1/3/2020 What are the most profound and important discoveries biologists have made? ■ 1) Darwin’s theory of Evolution by Natural Selection – Explains how species change over time Charles Darwin (1809-1889) Tree of Life 9 What are the most profound and important discoveries biologists have made? ■ 2) Watson & Crick (and others) discover that the heritable information evolution depends on is stored via DNA – A vast, incredibly complex (and beautiful) double helix structure ■ Allows information to be stored ■ Copied ■ Shuffled randomly ■ Changed (hopefully not too much!) Tree of Life 10 5 1/3/2020 What are the most profound and important discoveries biologists have made? ■ 3) The Tree of Life – The realization that all life on Earth is literally related ■ Every species on Earth shares a common ancestor with every other species! ■ You are literally, genealogically related not just to chimpanzees…. -

Phylogeny and the Tree of Life 537 That Pines and firs Are Different Enough to Be Placed in Sepa- History
how do biologists distinguish and categorize the millions of species on Earth? An understanding of evolutionary relationships suggests 26 one way to address these questions: We can decide in which “container” to place a species by comparing its traits with those of potential close relatives. For example, the scaly-foot does not have a fused eyelid, a highly mobile jaw, or a short tail posterior to the anus, three traits shared by all snakes. Phylogeny and These and other characteristics suggest that despite a superfi- cial resemblance, the scaly-foot is not a snake. Furthermore, a the Tree of Life survey of the lizards reveals that the scaly-foot is not alone; the legless condition has evolved independently in several different groups of lizards. Most legless lizards are burrowers or live in grasslands, and like snakes, these species lost their legs over generations as they adapted to their environments. Snakes and lizards are part of the continuum of life ex- tending from the earliest organisms to the great variety of species alive today. In this unit, we will survey this diversity and describe hypotheses regarding how it evolved. As we do so, our emphasis will shift from the process of evolution (the evolutionary mechanisms described in Unit Four) to its pattern (observations of evolution’s products over time). To set the stage for surveying life’s diversity, in this chapter we consider how biologists trace phylogeny, the evolution- ary history of a species or group of species. A phylogeny of lizards and snakes, for example, indicates that both the scaly- foot and snakes evolved from lizards with legs—but that they evolved from different lineages of legged lizards. -

Edvard Munch's to the Forest: Nature As Medium and Metaphor
Edvard Munch’s To the Forest : Nature as Medium and Metaphor Mara C. West A thesis submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Arts in the Department of Art. Chapel Hill 2007 Approved by: Mary D. Sheriff Dorothy Hoogland Verkerk Mary Pardo ABSTRACT MARA C. WEST: Edvard Munch’s To the Forest : Nature as Medium and Metaphor (Under the direction of Mary D. Sheriff) Edvard Munch, in his many paintings and prints, frequently utilizes biological concepts and botanical imagery as explanatory metaphors for human problems. Munch’s biological and natural motifs seem heavily indebted to nineteenth-century scientific and philosophical thinking, particularly Vitalism, and the ideas of Nietzsche. Munch’s woodcuts include some of his most interesting visualizations of the themes of biology; the woodcut To the Forest ( Mot Skogen ) is one such work in which biological concepts are central to its meaning. References to the natural world occur in both the representation of a forest, while its very medium is wood. The woodcut is thus a nexus where, in its medium and its message, nineteenth-century ideas about nature, science, love and the human soul, coexist in a struggle for coherence. ii Table of Contents I. Introduction ..................................................................................................................... 5 II. Nineteenth-Century Biological Metaphor and the “Berlin-bohème”........................... 13 III. Trees of Knowledge, -

Article Fast Track Tree of Life Reveals Clock-Like Speciation and Diversification Open Access
Tree of Life Reveals Clock-Like Speciation and Diversification S. Blair Hedges,*,1,2,3 Julie Marin,1,2,3 Michael Suleski,1,2,3 Madeline Paymer,1,2,3 and Sudhir Kumar1,2,3 1Center for Biodiversity, Temple University 2Institute for Genomics and Evolutionary Medicine, Temple University 3Department of Biology, Temple University *Corresponding author: E-mail: [email protected]. Associate editor: Emma Teeling Abstract Genomic data are rapidly resolving the tree of living species calibrated to time, the timetree of life, which will provide a framework for research in diverse fields of science. Previous analyses of taxonomically restricted timetrees have found a decline in the rate of diversification in many groups of organisms, often attributed to ecological interactions among species. Here, we have synthesized a global timetree of life from 2,274 studies representing 50,632 species and examined the pattern and rate of diversification as well as the timing of speciation. We found that species diversity has been mostly expanding overall and in many smaller groups of species, and that the rate of diversification in eukaryotes has been mostly constant. We also identified, and avoided, potential biases that may have influenced previous analyses of diver- sification including low levels of taxon sampling, small clade size, and the inclusion of stem branches in clade analyses. We found consistency in time-to-speciation among plants and animals, ~2 My, as measured by intervals of crown and stem species times. Together, this clock-like change at different levels suggests that speciation and diversification are processes dominated by random events and that adaptive change is largely a separate process.