Temporal Deregulation of Genes and Micrornas in Neurons During Prion-Induced Neurodegeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Causative Gene for Simmental Arachnomelia Syndrome Using a Network-Based Disease Gene Prioritization Approach
Identification of the Causative Gene for Simmental Arachnomelia Syndrome Using a Network-Based Disease Gene Prioritization Approach Shihui Jiao1., Qin Chu2., Yachun Wang1*, Zhenquan Xie3, Shiyu Hou3, Airong Liu5, Hongjun Wu4, Lin Liu6, Fanjun Geng7, Congyong Wang7, Chunhua Qin8, Rui Tan9, Xixia Huang10, Shixin Tan11, Meng Wu12, Xianzhou Xu12, Xuan Liu1, Ying Yu1, Yuan Zhang1 1 Key Laboratory of Agricultural Animal and Breeding, National Engineering Laboratory for Animal Breeding, College of Animal Science and Technology, China Agricultural University, Beijing, China, 2 Institute of Animal Husbandry and Veterinary Medicine, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China, 3 Anshan Hengli Dairy Farm, Anshan, Liaoning, China, 4 Xiertala Breeding Farm, Hailaer Farm Buro, Hailaer, Inner Mongolia, China, 5 Hailaer Farm Buro, Hailaer, Inner Mongolia, China, 6 Beijing Dairy Cattle Centre, Beijing, China, 7 Dingyuan Seedstock Bulls Breeding Ltd. Company, Zhengzhou, Henan, China, 8 Ningxia Sygen BioEngineering Research Center, Yinchuan, Ningxia, China, 9 Xinjiang General Livestock Service, Urumqi, Xinjiang, China, 10 College of Animal Science, Xinjiang Agriculture University, Urumqi, Xinjiang, China, 11 Xinjiang Tianshan Animal Husbandry Bio-engineering Co. Ltd, Urumqi, Xinjiang, China, 12 Dalian Xuelong Industry Limited Group, Dalian, Liaoning, China Abstract Arachnomelia syndrome (AS), mainly found in Brown Swiss and Simmental cattle, is a congenital lethal genetic malformation of the skeletal system. In this study, a network-based disease gene prioritization approach was implemented to rank genes in the previously reported ,7 Mb region on chromosome 23 associated with AS in Simmental cattle. The top 6 ranked candidate genes were sequenced in four German Simmental bulls, one known AS-carrier ROMEL and a pooled sample of three known non-carriers (BOSSAG, RIFURT and HIRMER). -
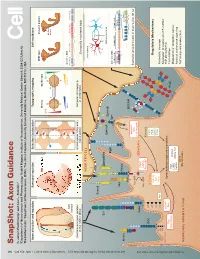
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Cytokine-Enhanced Cytolytic Activity of Exosomes from NK Cells
Cancer Gene Therapy https://doi.org/10.1038/s41417-021-00352-2 ARTICLE Cytokine-enhanced cytolytic activity of exosomes from NK Cells 1 1 2 3 2 3 Yutaka Enomoto ● Peng Li ● Lisa M. Jenkins ● Dimitrios Anastasakis ● Gaelyn C. Lyons ● Markus Hafner ● Warren J. Leonard 1 Received: 4 February 2021 / Revised: 9 May 2021 / Accepted: 18 May 2021 This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply 2021. This article is published with open access Abstract Natural killer (NK) cells play key roles in immune surveillance against tumors and viral infection. NK cells distinguish abnormal cells from healthy cells by cell–cell interaction with cell surface proteins and then attack target cells via multiple mechanisms. In addition, extracellular vesicles (EVs) derived from NK cells (NK-EVs), including exosomes, possess cytotoxic capacity against tumor cells, but their characteristics and regulation by cytokines remain unknown. Here, we report that EVs derived from human NK-92 cells stimulated with IL-15 + IL-21 show enhanced cytotoxic capacity against tumor cells. Major cytolytic granules, granzyme B and granzyme H, are enriched by IL-15 + IL-21 stimulation in NK-EVs; however, knockout experiments reveal those cytolytic granules are independent of enhanced cytotoxic capacity. To find out the key molecules, mass spectrometry analyses were 1234567890();,: 1234567890();,: performed with different cytokine conditions, no cytokine, IL-15, IL-21, or IL-15 + IL-21. We then found that CD226 (DNAM-1) on NK-EVs is enriched by IL-15 + IL-21 stimulation and that blocking antibodies against CD226 reduced the cytolytic activity of NK-EVs. -

Ck1δ Over-Expressing Mice Display ADHD-Like Behaviors, Frontostriatal Neuronal Abnormalities and Altered Expressions of ADHD-Candidate Genes
Molecular Psychiatry (2020) 25:3322–3336 https://doi.org/10.1038/s41380-018-0233-z ARTICLE CK1δ over-expressing mice display ADHD-like behaviors, frontostriatal neuronal abnormalities and altered expressions of ADHD-candidate genes 1 1 1 2 1 1 1 Mingming Zhou ● Jodi Gresack ● Jia Cheng ● Kunihiro Uryu ● Lars Brichta ● Paul Greengard ● Marc Flajolet Received: 8 November 2017 / Revised: 4 July 2018 / Accepted: 18 July 2018 / Published online: 19 October 2018 © Springer Nature Limited 2018 Abstract The cognitive mechanisms underlying attention-deficit hyperactivity disorder (ADHD), a highly heritable disorder with an array of candidate genes and unclear genetic architecture, remain poorly understood. We previously demonstrated that mice overexpressing CK1δ (CK1δ OE) in the forebrain show hyperactivity and ADHD-like pharmacological responses to D- amphetamine. Here, we demonstrate that CK1δ OE mice exhibit impaired visual attention and a lack of D-amphetamine- induced place preference, indicating a disruption of the dopamine-dependent reward pathway. We also demonstrate the presence of abnormalities in the frontostriatal circuitry, differences in synaptic ultra-structures by electron microscopy, as 1234567890();,: 1234567890();,: well as electrophysiological perturbations of both glutamatergic and GABAergic transmission, as observed by altered frequency and amplitude of mEPSCs and mIPSCs. Furthermore, gene expression profiling by next-generation sequencing alone, or in combination with bacTRAP technology to study specifically Drd1a versus Drd2 medium spiny neurons, revealed that developmental CK1δ OE alters transcriptional homeostasis in the striatum, including specific alterations in Drd1a versus Drd2 neurons. These results led us to perform a fine molecular characterization of targeted gene networks and pathway analysis. Importantly, a large fraction of 92 genes identified by GWAS studies as associated with ADHD in humans are significantly altered in our mouse model. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Xenopus Piwi Proteins Interact with a Broad Proportion of the Oocyte Transcriptome
Downloaded from rnajournal.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press Xenopus Piwi proteins interact with a broad proportion of the oocyte transcriptome JAMES A. TOOMBS,1,2,4 YULIYA A. SYTNIKOVA,3,5 GUNG-WEI CHIRN,3,6 IGNATIUS ANG,3,7 NELSON C. LAU,3 and MICHAEL D. BLOWER1,2 1Department of Molecular Biology, Massachusetts General Hospital, Boston, Massachusetts 02114, USA 2Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, USA 3Department of Biology and Rosenstiel Basic Medical Science Research Center, Brandeis University, Waltham, Massachusetts 02454, USA ABSTRACT Piwi proteins utilize small RNAs (piRNAs) to recognize target transcripts such as transposable elements (TE). However, extensive piRNA sequence diversity also suggests that Piwi/piRNA complexes interact with many transcripts beyond TEs. To determine Piwi target RNAs, we used ribonucleoprotein-immunoprecipitation (RIP) and cross-linking and immunoprecipitation (CLIP) to identify thousands of transcripts associated with the Piwi proteins XIWI and XILI (Piwi-protein-associated transcripts, PATs) from early stage oocytes of X. laevis and X. tropicalis. Most PATs associate with both XIWI and XILI and include transcripts of developmentally important proteins in oogenesis and embryogenesis. Only a minor fraction of PATs in both frog species displayed near perfect matches to piRNAs. Since predicting imperfect pairing between all piRNAs and target RNAs remains intractable, we instead determined that PAT read counts correlate well with the lengths and expression levels of transcripts, features that have also been observed for oocyte mRNAs associated with Drosophila Piwi proteins. We used an in vitro assay with exogenous RNA to confirm that XIWI associates with RNAs in a length- and concentration-dependent manner. -

Supporting Information for Proteomics DOI 10.1002/Pmic.200400942
Supporting Information for Proteomics DOI 10.1002/pmic.200400942 Wei-Jun Qian, Jon M. Jacobs, David G. Camp II, Matthew E. Monroe, Ronald J. Moore, Marina A. Gritsenko, Steve E. Calvano, Stephen F. Lowry, Wenzhong Xiao, Lyle L. Moldawer, Ronald W. Davis, Ronald G. Tompkins and Richard D. Smith Comparative proteome analyses of human plasma following in vivo lipopolysaccharide administration using multidimensional separations coupled with tandem mass spectrometry ã 2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.de Supplemental Table 1: List of 804 identified plasma proteins. Note: Reference IDs correspond to either SwissProt, NCBI, or PIR database entries. For each protein, one representive peptide sequence and the peptide charge_state, SEQUEST Xcorr, and Delcn are listed. The number of different peptides identifying each specific protein is also indicated. To facilitate comparison of protein abundances between the untreated and treated samples, the numbers of peptide hit, the protein abundance ratio (treated/untreated) calculated from peptide peak area ratios, and the standard deviation of peptide peak area ratios for each protein are also listed. No abundance ratio is shown if the protein has no common peptide identified in the two samples. Whether the same protein ID was reported in reference 16 and reference 27 (195 proteins observed from two different sources) is also indicated. Reference Description # of different Representitive peptide Charge_state Xcorr DelCn peptides Peptide Hits Peptide Protein Standard -

The Tumor Suppressor Notch Inhibits Head and Neck Squamous Cell
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2015 THE TUMOR SUPPRESSOR NOTCH INHIBITS HEAD AND NECK SQUAMOUS CELL CARCINOMA (HNSCC) TUMOR GROWTH AND PROGRESSION BY MODULATING PROTO-ONCOGENES AXL AND CTNNAL1 (α-CATULIN) Shhyam Moorthy Shhyam Moorthy Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Biochemistry, Biophysics, and Structural Biology Commons, Cancer Biology Commons, Cell Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Moorthy, Shhyam and Moorthy, Shhyam, "THE TUMOR SUPPRESSOR NOTCH INHIBITS HEAD AND NECK SQUAMOUS CELL CARCINOMA (HNSCC) TUMOR GROWTH AND PROGRESSION BY MODULATING PROTO-ONCOGENES AXL AND CTNNAL1 (α-CATULIN)" (2015). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 638. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/638 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. THE TUMOR SUPPRESSOR NOTCH INHIBITS HEAD AND NECK SQUAMOUS CELL CARCINOMA (HNSCC) TUMOR GROWTH AND PROGRESSION BY MODULATING PROTO-ONCOGENES AXL AND CTNNAL1 (α-CATULIN) by Shhyam Moorthy, B.S. -

Signal Transduction in L-DOPA-Induced Dyskinesia: from Receptor Sensitization to Abnormal… Et Al
Journal of Neural Transmission https://doi.org/10.1007/s00702-018-1847-7 NEUROLOGY AND PRECLINICAL NEUROLOGICAL STUDIES - REVIEW ARTICLE Signal transduction in L‑DOPA‑induced dyskinesia: from receptor sensitization to abnormal gene expression Giada Spigolon1 · Gilberto Fisone1 Received: 1 November 2017 / Accepted: 23 January 2018 © The Author(s) 2018. This article is an open access publication Abstract A large number of signaling abnormalities have been implicated in the emergence and expression of L-DOPA-induced dyskinesia (LID). The primary cause for many of these changes is the development of sensitization at dopamine receptors located on striatal projection neurons (SPN). This initial priming, which is particularly evident at the level of dopamine D1 receptors (D1R), can be viewed as a homeostatic response to dopamine depletion and is further exacerbated by chronic administration of L-DOPA, through a variety of mechanisms afecting various components of the G-protein-coupled receptor machinery. Sensitization of dopamine receptors in combination with pulsatile administration of L-DOPA leads to intermit- tent and coordinated hyperactivation of signal transduction cascades, ultimately resulting in long-term modifcations of gene expression and protein synthesis. A detailed mapping of these pathological changes and of their involvement in LID has been produced during the last decade. According to this emerging picture, activation of sensitized D1R results in the stimulation of cAMP-dependent protein kinase and of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa. This, in turn, activates the extracellular signal-regulated kinases 1 and 2 (ERK), leading to chromatin remodeling and aberrant gene transcription. Dysregulated ERK results also in the stimulation of the mammalian target of rapamycin complex 1, which promotes protein synthesis.