Regulation of Glucose Metabolism in Muscles from Dexamethasone Treated Rats
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Statistical Analysis Plan (SAP)
Statistical Analysis Plan (SAP) Protocol Title: A Phase IIa, two-part, randomised, multi-centre, multinational, double-blind, placebo-controlled, parallel group study to compare the efficacy and safety of BCT197 when added on to standard of care for the treatment of acute respiratory exacerbations of chronic obstructive pulmonary disease (COPD) requiring hospitalisation in adults Protocol Number: MBCT206 Protocol Version, Date Final V1.0, 05 November 2015 Final V2.0 (Amendment 1), 23 December 2015 Final V3.0 (Amendment 2), 10 March 2016 Final V4.0 (Amendment 3), 26 May 2016 Final V5.0 (Amendment 4), 08 November 2016 ICON ID: 3082/0004 Document Version, Date: FINAL 1.0, 07 September 2017 FINAL 2.0, 16 November 2017 Prepared by: ICON Clinical Research Services On behalf of: Mereo BioPharma 1 Ltd Statistical Analysis Plan Version: FINAL 2.0, Date:16 November 2017 Protocol Number: MBCT206 Page 1 of 112 Statistical Analysis Plan (SAP) Confidentiality statement: The information provided in this document is strictly confidential. The recipients of the SAP must not disclose the confidential information . contained within this document or any related information to other persons without the permission of the sponsor. In addition, the recipients of the SAP must keep this confidential document in a . controlled environment which prevents unauthorized access to the document. Statistical Analysis Plan Version: FINAL 2.0, Date:16 November 2017 Protocol Number: MBCT206 Page 2 of 112 Statistical Analysis Plan (SAP) SIGNATURE PAGE Prepared at ICON Clinical Research by: Date (DD Mmm YYYY) Reviewed at ICON Clinical Research by: Date (DD Mmm YYYY) Approved at Mereo BioPharma 1 Ltd by: Date (DD Mmm YYYY) Statistical Analysis Plan Version: FINAL 2.0, Date:16 November 2017 Protocol Number: MBCT206 Page 3 of 112 Statistical Analysis Plan (SAP) REVISION HISTORY Version/Date Version name Section Changes implemented Version 1.0/ Initial approved N/A N/A 07-Sep-2017 version 7.1.4.13 Clinical treatment failure Medical review clarification added. -

Evaluating Onco-Geriatric Scores and Medication Risks to Improve Cancer Care for Older Patients
Evaluating onco-geriatric scores and medication risks to improve cancer care for older patients Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn vorgelegt von IMKE ORTLAND aus Quakenbrück Bonn 2019 Angefertigt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn. Diese Dissertation ist auf dem Hochschulschriftenserver der ULB Bonn elektronisch publiziert. https://nbn-resolving.org/urn:nbn:de:hbz:5-58042 Erstgutachter: Prof. Dr. Ulrich Jaehde Zweitgutachter: Prof. Dr. Andreas Jacobs Tag der Promotion: 28. Februar 2020 Erscheinungsjahr: 2020 Danksagung Auf dem Weg zur Promotion haben mich viele Menschen begleitet und in ganz unterschiedlicher Weise unterstützt. All diesen Menschen möchte ich an dieser Stelle ganz herzlich danken. Mein aufrichtiger Dank gilt meinem Doktorvater Prof. Dr. Ulrich Jaehde für das in mich gesetzte Vertrauen, sowie für die Überlassung dieses spannenden Dissertationsthemas. Die uneingeschränkte Unterstützung, wertvollen Diskussionen und die mitreißende Begeisterung für die Wissenschaft haben mich während aller Phasen der Dissertation stets motiviert, unterstützt und sehr viel Wertvolles gelehrt. Prof. Dr. Andreas Jacobs danke ich herzlich für die Initiierung dieses interessanten Projekts, für die stetige Begeisterung und Unterstützung, sowie für das mir entgegengebrachte Vertrauen. Die ausgezeichnete Zusammenarbeit mit dem Johanniter Krankenhaus Bonn hat ganz maßgeblich zum Gelingen dieser Arbeit beigetragen. Auch danke ich Prof. Dr. Andreas Jacobs herzlich für die Bereitschaft, das Koreferat dieser Arbeit zu übernehmen. Ebenfalls danke ich herzlich Prof. Dr. Yon-Dschun Ko für seine fortwährende Motivation und seinen Einsatz, sowie für das mir geschenkte Vertrauen, dieses Projekt am Johanniter Krankenhaus zu realisieren. Ebenfalls danke ich Prof. -

Medication Risks in Older Patients with Cancer
Medication risks in older patients with cancer 1 Medication risks in older patients (70+) with cancer and their association with therapy-related toxicity Imke Ortland1, Monique Mendel Ott1, Michael Kowar2, Christoph Sippel3, Yon-Dschun Ko3#, Andreas H. Jacobs2#, Ulrich Jaehde1# 1 Institute of Pharmacy, Department of Clinical Pharmacy, University of Bonn, An der Immenburg 4, 53121 Bonn, Germany 2 Department of Geriatrics and Neurology, Johanniter Hospital Bonn, Johanniterstr. 1-3, 53113 Bonn, Germany 3 Department of Oncology and Hematology, Johanniter Hospital Bonn, Johanniterstr. 1-3, 53113 Bonn, Germany # equal contribution Corresponding author Ulrich Jaehde Institute of Pharmacy University of Bonn An der Immenburg 4 53121 Bonn, Germany Phone: +49 228-73-5252 Fax: +49-228-73-9757 [email protected] Medication risks in older patients with cancer 2 Abstract Objectives To evaluate medication-related risks in older patients with cancer and their association with severe toxicity during antineoplastic therapy. Methods This is a secondary analysis of two prospective, single-center observational studies which included patients ≥ 70 years with cancer. The patients’ medication was investigated regarding possible risks: polymedication (defined as the use of ≥ 5 drugs), potentially inadequate medication (PIM; defined by the EU(7)-PIM list), and relevant potential drug- drug interactions (rPDDI; analyzed by the ABDA interaction database). The risks were analyzed at two different time points: before and after start of cancer therapy. Severe toxicity during antineoplastic therapy was captured from medical records according to the Common Terminology Criteria for Adverse Events (CTCAE). The association between Grade ≥ 3 toxicity and medication risks was evaluated by univariate regression. -
PASS Information
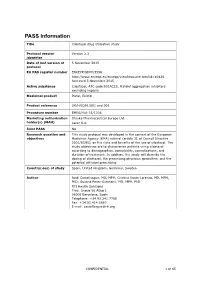
PASS Information Title Cilostazol drug utilisation study Protocol version Version 2.3 identifier Date of last version of 5 November 2015 protocol EU PAS register number ENCEPP/SDPP/3596 http://www.encepp.eu/encepp/viewResource.htm?id=10426 Accessed 5 November 2015 Active substance Cilostazol, ATC code B01AC23, Platelet aggregation inhibitors excluding heparin Medicinal product Pletal, Ekistol Product reference UK/H/0291/001 and 002 Procedure number EMEA/H/A-31/1306 Marketing authorisation Otsuka Pharmaceutical Europe Ltd. holder(s) (MAH) Lacer S.A. Joint PASS No Research question and This study protocol was developed in the context of the European objectives Medicines Agency (EMA) referral (article 31 of Council Directive 2001/83/EC) on the risks and benefits of the use of cilostazol. The study objectives are to characterise patients using cilostazol according to demographics, comorbidity, comedications, and duration of treatment. In addition, the study will describe the dosing of cilostazol, the prescribing physician specialties, and the potential off-label prescribing. Country(-ies) of study Spain, United Kingdom, Germany, Sweden Author Jordi Castellsague, MD, MPH; Cristina Varas-Lorenzo, MD, MPH, PhD; Susana Perez-Gutthann, MD, MPH, PhD RTI Health Solutions Trav. Gracia 56 Atico 1 08006 Barcelona, Spain Telephone: +34.93.241.7766 Fax: +34.93.414.2610 E-mail: [email protected] CONFIDENTIAL 1 of 65 Marketing Authorisation Holder(s) Marketing authorisation Otsuka Pharmaceutical Europe Ltd. holder(s) Gallions Wexham Springs Framewood Road Wexham SL3 6PJ, UK Lacer S.A. Sardenya 350 08025 Barcelona Spain MAH contact person Dr Marco Avila Regional Vice President, Medical Europe Otsuka Pharmaceutical Europe Ltd EU QPPV Dr Achint Kumar Otsuka Europe Development and Commercialisation Limited (OEDC) Head of Medical Dr Sanjay Kapoor Compliance-Europe Otsuka Pharmaceutical Europe Ltd. -

Estonian Statistics on Medicines 2016 1/41
Estonian Statistics on Medicines 2016 ATC code ATC group / Active substance (rout of admin.) Quantity sold Unit DDD Unit DDD/1000/ day A ALIMENTARY TRACT AND METABOLISM 167,8985 A01 STOMATOLOGICAL PREPARATIONS 0,0738 A01A STOMATOLOGICAL PREPARATIONS 0,0738 A01AB Antiinfectives and antiseptics for local oral treatment 0,0738 A01AB09 Miconazole (O) 7088 g 0,2 g 0,0738 A01AB12 Hexetidine (O) 1951200 ml A01AB81 Neomycin+ Benzocaine (dental) 30200 pieces A01AB82 Demeclocycline+ Triamcinolone (dental) 680 g A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+ Thymol (dental) 3094 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+ Cetylpyridinium chloride (gingival) 227150 g A01AD81 Lidocaine+ Cetrimide (O) 30900 g A01AD82 Choline salicylate (O) 864720 pieces A01AD83 Lidocaine+ Chamomille extract (O) 370080 g A01AD90 Lidocaine+ Paraformaldehyde (dental) 405 g A02 DRUGS FOR ACID RELATED DISORDERS 47,1312 A02A ANTACIDS 1,0133 Combinations and complexes of aluminium, calcium and A02AD 1,0133 magnesium compounds A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 811120 pieces 10 pieces 0,1689 A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 3101974 ml 50 ml 0,1292 A02AD83 Calcium carbonate+ Magnesium carbonate (O) 3434232 pieces 10 pieces 0,7152 DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 46,1179 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 2,3855 A02BA02 Ranitidine (O) 340327,5 g 0,3 g 2,3624 A02BA02 Ranitidine (P) 3318,25 g 0,3 g 0,0230 A02BC Proton pump inhibitors 43,7324 A02BC01 Omeprazole -

Statistical Analysis Plan I8K-MC-JPDA (Version 3)
Statistical Analysis Plan I8K-MC-JPDA (Version 3) A Randomized, Double-Blind, Placebo-Controlled, 2-Part Phase 2 Study to Evaluate the Safety and Efficacy of LY3337641 in Adult Subjects with Rheumatoid Arthritis: The RAjuvenate Study NCT02628028 Approval Date: 01-Sep-2017 I8K-MC-JPDA Statistical Analysis Plan Version 3 Page 1 1. Statistical Analysis Plan: I8K-MC-JPDA: A Randomized, Double-Blind, Placebo- Controlled, 2-Part Phase 2 Study to Evaluate the Safety and Efficacy of LY3337641 in Adult Subjects with Rheumatoid Arthritis: The RAjuvenate Study Confidential Information The information contained in this Statistical Analysis Plan (SAP) is confidential and the information contained within it may not be reproduced or otherwise disseminated without the approval of Eli Lilly and Company or its subsidiaries. This document and its associated attachments or appendices are subject to United States Freedom of Information Act Exemption 4. LY3337641 Rheumatoid Arthritis Study I8K-MC-JPDA is a 2-part Phase 2, randomized, double-blind, placebo-controlled trial in subjects with rheumatoid arthritis. In Part A, subjects will be dosed with 5, 10, or 30 mg of LY3337641 or placebo for 4 weeks. Following a safety analysis, Part B will enroll subjects to be dosed with the planned doses of 5, 10, or 30 mg of LY3337641 or placebo for 12 weeks. Eli Lilly and Company Indianapolis, Indiana USA 46285 Protocol I8K-MC-JPDA Phase 2 Statistical Analysis Plan electronically signed and approved by Lilly: 14 September 2016 SAP Version 2 electronically signed and approved by Lilly on date provided below. 01 March 2017 SAP Version 3 electronically signed and approved by Lilly on date provided below. -

Controlled, Parallel Group Phase 2A Study to Assess the Efficacy of Ro5459072 in Patients with Primary Sjögren’S Syndrome
Official Title: A MULTI-CENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO- CONTROLLED, PARALLEL GROUP PHASE 2A STUDY TO ASSESS THE EFFICACY OF RO5459072 IN PATIENTS WITH PRIMARY SJÖGREN’S SYNDROME NCT Number: NCT02701985 Document Dates: Protocol Version 3: 30-Oct-2016 PROTOCOL TITLE: A MULTI-CENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED, PARALLEL GROUP PHASE 2A STUDY TO ASSESS THE EFFICACY OF RO5459072 IN PATIENTS WITH PRIMARY SJÖGREN’S SYNDROME PROTOCOL NUMBER: BP30037 VERSION: 3 EUDRACT NUMBER: 2015-004476-30 IND NUMBER: 128528 TEST PRODUCT: RO5459072 SPONSOR: F. Hoffmann-La Roche Ltd DATE FINAL: Version 1: 28 January 2016 DATE AMENDED Version 2: 01 August 2016 Version 3: See electronic date stamp below FINAL PROTOCOL APPROVAL Approver's Name Title Date and Time (UTC) Translational Medicine Leader 30-Oct-2016 15:40:47 CONFIDENTIAL STATEMENT The information contained in this document, especially any unpublished data, is the property of F.Hoffmann-La Roche Ltd (or under its control) and therefore is provided to you in confidence as an investigator, potential investigator, or consultant, for review by you, your staff, and an applicable Ethics Committee or Institutional Review Board. It is understood that this information will not be disclosed to others without written authorization from Roche except to the extent necessary to obtain informed consent from persons to whom the drug may be administered. RO5459072— F. Hoffmann-La Roche Ltd Protocol BP30037 Version 3 PROTOCOL AMENDMENT, VERSION 3: RATIONALE Protocol BP30037 has been amended to incorporate the following changes to the protocol: Section 4.2.3: Implementation of changes to eligibility criteria . The eligibility criteria of the protocol have therefore been amended to mandate testing for tuberculosis and exclude patients with positive results. -

Estonian Statistics on Medicines 2013 1/44
Estonian Statistics on Medicines 2013 DDD/1000/ ATC code ATC group / INN (rout of admin.) Quantity sold Unit DDD Unit day A ALIMENTARY TRACT AND METABOLISM 146,8152 A01 STOMATOLOGICAL PREPARATIONS 0,0760 A01A STOMATOLOGICAL PREPARATIONS 0,0760 A01AB Antiinfectives and antiseptics for local oral treatment 0,0760 A01AB09 Miconazole(O) 7139,2 g 0,2 g 0,0760 A01AB12 Hexetidine(O) 1541120 ml A01AB81 Neomycin+Benzocaine(C) 23900 pieces A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+Thymol(dental) 2639 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+Cetylpyridinium chloride(gingival) 179340 g A01AD81 Lidocaine+Cetrimide(O) 23565 g A01AD82 Choline salicylate(O) 824240 pieces A01AD83 Lidocaine+Chamomille extract(O) 317140 g A01AD86 Lidocaine+Eugenol(gingival) 1128 g A02 DRUGS FOR ACID RELATED DISORDERS 35,6598 A02A ANTACIDS 0,9596 Combinations and complexes of aluminium, calcium and A02AD 0,9596 magnesium compounds A02AD81 Aluminium hydroxide+Magnesium hydroxide(O) 591680 pieces 10 pieces 0,1261 A02AD81 Aluminium hydroxide+Magnesium hydroxide(O) 1998558 ml 50 ml 0,0852 A02AD82 Aluminium aminoacetate+Magnesium oxide(O) 463540 pieces 10 pieces 0,0988 A02AD83 Calcium carbonate+Magnesium carbonate(O) 3049560 pieces 10 pieces 0,6497 A02AF Antacids with antiflatulents Aluminium hydroxide+Magnesium A02AF80 1000790 ml hydroxide+Simeticone(O) DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 34,7001 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 3,5364 A02BA02 Ranitidine(O) 494352,3 g 0,3 g 3,5106 A02BA02 Ranitidine(P) -

Statistical Analysis Plan
Cover Page Official title: A randomised, double blind, placebo controlled trial to evaluate the effect of tralokinumab on vaccine antibody responses in adults with moderate-to-severe atopic dermatitis who are candidates for systemic therapy LEO Pharma number: LP0162-1341 NCT number: NCT03562377 Date: 18-Oct-2019 Statistical analysis plan Vaccine responses in tralokinumab-treated atopic dermatitis ECZTRA 5 (ECZema TRAlokinumab trial no. 5) Phase 2 – Vaccine response trial A randomised, double-blind, placebo-controlled trial to evaluate the effect of tralokinumab on vaccine antibody responses in adults with moderate-to-severe atopic dermatitis who are candidates for systemic therapy LEO Pharma A/S Trial ID: LP0162-1341 Date: 18-OCT-2019 Version: FINAL TMF-000095060 - Version 1. 0 1 Statistical analysis plan approval 1.1 Approval statement On behalf of LEO, the Biostatistics Lead and the Medical Lead, are authorised to approve the statistical analysis plan. The QC statistician has by approving this document confirmed that the statistical information has been subject to statistical quality control. The following persons have approved this statistical analysis plan using electronic signatures as presented on the last page of this document. PPD Biostatistics Lead, Medical Sciences PPD Medical Lead, Medical Sciences PPD QC statistician, Biostatistics TMF-000095060 - Version 1. 0 2 Statistical analysis plan statements 2.1 Compliance with Good Clinical Practice This statistical analysis plan is designed to comply with the standards issued by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) (E3: Structure and Content of Clinical Study Reports, E6: Good Clinical Practice, and E9: Statistical Principles for Clinical Trials). -

The Selection and Use of Essential Medicines
WHO Technical Report Series 958 THE SELECTION AND USE OF ESSENTIAL MEDICINES This report presents the recommendations of the WHO Expert THE SELECTION AND USE Committee responsible for updating the WHO Model List of Essential Medicines. The fi rst part contains a review of the OF ESSENTIAL MEDICINES report of the meeting of the Expert Subcommittee on the Selection and Use of Essential Medicines, held in October 2008. It also provides details of new applications for paediatric medicines and summarizes the Committee’s considerations and justifi cations for additions and changes to the Model List, including its recommendations. Part Two of the publication is the report of the second meeting of the Subcommittee of the Expert Committee on the Selection and Use of Essential Medicines. Annexes include the revised version of the WHO Model List of Essential Medicines (the 16th) and the revised version of the WHO Model List of Report of the WHO Expert Committee, 2009 Essential Medicines for Children (the 2nd). In addition there is a list of all the items on the Model List sorted according to their (including the 16th WHO Model List of Essential Medicines Anatomical Therapeutic Chemical (ATC) classifi cation codes. and the 2nd WHO Model List of Essential Medicines for Children) WHO Technical Report Series — 958 WHO Technical ISBN 978-92-4-120958-8 Geneva TTRS958cover.inddRS958cover.indd 1 110.06.100.06.10 008:328:32 The World Health Organization was established in 1948 as a specialized agency of the United Nations serving as the directing and coordinating authority for SELECTED WHO PUBLICATIONS OF RELATED INTEREST international health matters and public health. -

Review of Existing Classification Efforts
Project No. TREN-05-FP6TR-S07.61320-518404-DRUID DRUID Driving under the Influence of Drugs, Alcohol and Medicines Integrated Project 1.6. Sustainable Development, Global Change and Ecosystem 1.6.2: Sustainable Surface Transport 6th Framework Programme Deliverable 4.1.1 Review of existing classification efforts Due date of deliverable: (15.01.2008) Actual submission date: (07.02.2008) Start date of project: 15.10.2006 Duration: 48 months Organisation name of lead contractor for this deliverable: UGent Revision 1.0 Project co-funded by the European Commission within the Sixth Framework Programme (2002-2006) Dissemination Level PU Public X PP Restricted to other programme participants (including the Commission Services) RE Restricted to a group specified by the consortium (including the Commission Services) CO Confidential, only for members of the consortium (including the Commission Services) Task 4.1 : Review of existing classification efforts Authors: Kristof Pil, Elke Raes, Thomas Van den Neste, An-Sofie Goessaert, Jolien Veramme, Alain Verstraete (Ghent University, Belgium) Partners: - F. Javier Alvarez (work package leader), M. Trinidad Gómez-Talegón, Inmaculada Fierro (University of Valladolid, Spain) - Monica Colas, Juan Carlos Gonzalez-Luque (DGT, Spain) - Han de Gier, Sylvia Hummel, Sholeh Mobaser (University of Groningen, the Netherlands) - Martina Albrecht, Michael Heiβing (Bundesanstalt für Straßenwesen, Germany) - Michel Mallaret, Charles Mercier-Guyon (University of Grenoble, Centre Regional de Pharmacovigilance, France) - Vassilis Papakostopoulos, Villy Portouli, Andriani Mousadakou (Centre for Research and Technology Hellas, Greece) DRUID 6th Framework Programme Deliverable D.4.1.1. Revision 1.0 Review of Existing Classification Efforts Page 2 of 127 Introduction DRUID work package 4 focusses on the classification and labeling of medicinal drugs according to their influence on driving performance. -

COMPARISON of the WHO ATC CLASSIFICATION & Ephmra/Intellus Worldwide ANATOMICAL CLASSIFICATION
COMPARISON OF THE WHO ATC CLASSIFICATION & EphMRA/Intellus Worldwide ANATOMICAL CLASSIFICATION November 2020 Comparison of the WHO ATC Classification and EphMRA / Intellus Worldwide Anatomical Classification The following booklet is designed to improve the understanding of the two classification systems. The development of the two systems had previously taken place separately. EphMRA and WHO are now working together to ensure that there is a convergence of the 2 systems rather than a divergence. In order to better understand the two classification systems, we should pay attention to the way in which substances/products are classified. WHO mainly classifies substances according to the therapeutic or pharmaceutical aspects and in one class only (particular formulations or strengths can be given separate codes, e.g. clonidine in C02A as antihypertensive agent, N02C as anti-migraine product and S01E as ophthalmic product). EphMRA classifies products, mainly according to their indications and use. Therefore, it is possible to find the same compound in several classes, depending on the product, e.g., NAPROXEN tablets can be classified in M1A (antirheumatic), N2B (analgesic) and G2C if indicated for gynaecological conditions only. The purposes of classification are also different: The main purpose of the WHO classification is for international drug utilisation research and for adverse drug reaction monitoring. This classification is recommended by the WHO for use in international drug utilisation research. The EphMRA/Intellus Worldwide classification has a primary objective to satisfy the marketing needs of the pharmaceutical companies. Therefore, a direct comparison is sometimes difficult due to the different nature and purpose of the two systems. The aim of harmonisation is to reach a “full” agreement of all mono substances in a given class as listed in the WHO ATC Index, mainly at third level: whenever this is not possible, or harmonisation of third level is too difficult or makes no sense (e.g.