Research Article Nonhuman Primate Model of Oculocutaneous Albinism with TYR and OCA2 Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ophthalmology
Ophthalmology Information for health professionals MEDICAL GENETIC TESTING FOR OPHTHALMOLOGY Recent technologies, in particularly Next Generation Sequencing (NGS), allows fast, accurate and valuable diagnostic tests. For Ophthalmology, CGC Genetics has an extensive list of medical genetic tests with clinical integration of results by our Medical Geneticists. 1. EXOME SEQUENCING: Exome Sequencing is a very efficient strategy to study most exons of a patient’s genome, unraveling mutations associated with specific disorders or phenotypes. With this diagnostic strategy, patients can be studied with a significantly reduced turnaround time and cost. CGC Genetics has available 2 options for Exome Sequencing: • Whole Exome Sequencing (WES), which analyzes the entire exome (about 20 000 genes); • Disease Exome by CGC Genetics, which analyzes about 6 000 clinically-relevant genes. Any of these can be performed in the index case or in a Trio. 2. NGS PANELS For NGS panels, several genes associated with the same phenotype are simultaneously sequenced. These panels provide increased diagnostic capability with a significantly reduced turnaround time and cost. CGC Genetics has several NGS panels for Ophthalmology that are constantly updated (www.cgcgenetics.com). Any gene studied in exome or NGS panel can also be individually sequenced and analyzed for deletion/duplication events. 3. EXPERTISE IN MEDICAL GENETICS CGC Genetics has Medical Geneticists specialized in genetic counseling for ophthalmological diseases who may advice in choosing the most appropriate -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Albinism: Modern Molecular Diagnosis
British Journal of Ophthalmology 1998;82:189–195 189 Br J Ophthalmol: first published as 10.1136/bjo.82.2.189 on 1 February 1998. Downloaded from PERSPECTIVE Albinism: modern molecular diagnosis Susan M Carden, Raymond E Boissy, Pamela J Schoettker, William V Good Albinism is no longer a clinical diagnosis. The past cytes and into which melanin is confined. In the skin, the classification of albinism was predicated on phenotypic melanosome is later transferred from the melanocyte to the expression, but now molecular biology has defined the surrounding keratinocytes. The melanosome precursor condition more accurately. With recent advances in arises from the smooth endoplasmic reticulum. Tyrosinase molecular research, it is possible to diagnose many of the and other enzymes regulating melanin synthesis are various albinism conditions on the basis of genetic produced in the rough endoplasmic reticulum, matured in causation. This article seeks to review the current state of the Golgi apparatus, and translocated to the melanosome knowledge of albinism and associated disorders of hypo- where melanin biosynthesis occurs. pigmentation. Tyrosinase is a copper containing, monophenol, mono- The term albinism (L albus, white) encompasses geneti- oxygenase enzyme that has long been known to have a cally determined diseases that involve a disorder of the critical role in melanogenesis.5 It catalyses three reactions melanin system. Each condition of albinism is due to a in the melanin pathway. The rate limiting step is the genetic mutation on a diVerent chromosome. The cutane- hydroxylation of tyrosine into dihydroxyphenylalanine ous hypopigmentation in albinism ranges from complete (DOPA) by tyrosinase, but tyrosinase does not act alone. -
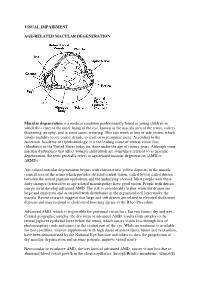
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

The Ocular Albinism Type 1 (OA1) GPCR Is Ubiquitinated and Its Traffic Requires Endosomal Sorting Complex Responsible for Transport (ESCRT) Function
The ocular albinism type 1 (OA1) GPCR is ubiquitinated and its traffic requires endosomal sorting complex responsible for transport (ESCRT) function Francesca Giordanoa,b,c,1, Sabrina Simoesa,b,c, and Graça Raposoa,b,c,2 aInstitut Curie, Centre de Recherche, Paris F-75248, France; bStructure and Membrane Compartments, Centre National de la Recherche Scientifique, UMR144, Paris F-75248, France; and cCell and Tissue Imaging Facility, Infrastructures en Biologie Sante et Agronomie, Paris F-75248, France Edited by David D. Sabatini, New York University School of Medicine, New York, NY, and approved June 7, 2011 (received for review March 2, 2011) The function of signaling receptors is tightly controlled by their tion and function remain undefined. Posttranslational modification intracellular trafficking. One major regulatory mechanism within the by ubiquitination controls intracellular trafficking events within fi endo-lysosomal system required for receptor localization and down- the endo-lysosomal system (10, 11). Ubiquitin-modi ed mem- regulation is protein modification by ubiquitination and down- brane proteins delivered from the endocytic or biosynthetic stream interactions with the endosomal sorting complex responsible pathways can be recognized by components of the endosomal sorting complex responsible for transport (ESCRT) machinery for transport (ESCRT) machinery. Whether and how these mecha- for targeting to intraluminal vesicles (ILVs) of multivesicular nisms operate to regulate endosomal sorting of mammalian G bodies (MVBs) and for subsequent -

Abnormal Foveal Morphology in Ocular Albinism Imaged with Spectral-Domain Optical Coherence Tomography
CLINICAL SCIENCES Abnormal Foveal Morphology in Ocular Albinism Imaged With Spectral-Domain Optical Coherence Tomography Gabriel T. Chong, MD; Sina Farsiu, PhD; Sharon F. Freedman, MD; Neeru Sarin, MBBS; Anjum F. Koreishi, BS; Joseph A. Izatt, PhD; Cynthia A. Toth, MD Objectives: To evaluate the spectrum of foveal archi- Pudlak syndrome: persistence of an abnormal, highly tecture in pediatric albinism and to assess the utility of reflective band across the fovea, multiple inner retinal spectral-domain optical coherence tomography (OCT) layers normally absent at the center of the fovea, and loss in ocular imaging of children with nystagmus. of the normally thickened photoreceptor nuclear layer at the fovea when compared with that in control sub- Methods: Spectral-domain OCT imaging was per- jects. The optic nerve was elevated in multiple eyes formed on study subjects in 3 groups: subjects with of subjects with OA or suspected OA and the subject ocular albinism (OA) or suspected OA with foveal hypo- with oculocutaneous albinism and Hermansky-Pudlak plasia, with nystagmus, and with or without iris trans- syndrome. illumination; a subject with oculocutaneous albinism and Hermansky-Pudlak syndrome; and control subjects. Dense Conclusions: A spectrum of foveal morphological ab- volumetric scans of each fovea were captured using stan- normalities is seen in subjects with OA or suspected OA, dard and handheld spectral-domain OCT devices. Im- which in some cases contrasted with previous studies ages were postprocessed and scored for the presence and using time-domain OCT. These OCT findings clarify configuration of each retinal layer across the fovea. the morphology of foveal hypoplasia seen clinically. -

Visual Consequences of Albinism
Bachelorarbeit des Studiengangs Augenoptik und Hörakustik Katharina Breher Visual Consequences Of Albinism Prüfer: Vertr.prof. Dr. Steffen Kreikemeier (Hochschule Aalen, Deutschland) Zweitprüfer: Dr. Willard Bleything, Distinguished University Professor of Optometry and Public Health (College of Optometry, Pacific University, Oregon, USA) Erklärung- I - Visual Consequences Of Albinism Zugelassene Abschlussarbeit des Studiengangs Augenoptik und Hörakustik zur Erlangung des akademischen Grades Bachelor of Science vorgelegt von Katharina Breher Tag der Einreichung: 25. Februar 2015 Fakultät Augenoptik und Hörakustik Hochschule Aalen Erklärung - II - Erklärung Ich versichere hiermit, dass ich die vorliegende Bachelorthesis selbstständig und ohne fremde Hilfe angefertigt und keine andere als die angegebene Literatur benutzt habe. Alle von anderen Autoren wörtlich übernommenen Stellen wie auch die sich an die Gedankengänge anderer Autoren eng anlehnenden Ausführungen meiner Arbeit sind besonders gekennzeichnet. Diese Arbeit wurde bisher in gleicher oder ähnlicher Form keiner anderen Prüfungsbehörde vorgelegt und auch nicht veröffentlicht. Ort, Datum Unterschrift Table of contents - 3 - Table of contents Erklärung ....................................................................................................... II Table of contents ........................................................................................... 3 Abstract ........................................................................................................ -

X-Linked Ocular Albinism: Prevalence and Mutations – a National Study
European Journal of Human Genetics (1998) 6, 570–577 t © 1998 Stockton Press All rights reserved 1018–4813/98 $12.00 http://www.stockton-press.co.uk/ejhg ORIGINAL PAPER X-linked ocular albinism: prevalence and mutations – a national study Thomas Rosenberg1 and Marianne Schwartz2 1National Eye Clinic for the Visually Impaired, Hellerup, Denmark 2Department of Clinical Genetics, The Juliane Marie Centre, Rigshospitalet, University Hospital, Copenhagen, Denmark In a national retrospective register study 112 patients with ocular albinism (OA) were identified, including 60 male patients with proven or presumed X-linked ocular albinism (XLOA). Based on the birth year cohorts 1960–1989, an XLOA point prevalence at birth of 1 in 60 000 live-born was calculated. We identified 14 XLOA families in the Danish population, and obtained DNA from affected persons in nine families. Mutation analysis of the OA1 gene demonstrated seven presumed pathogenic mutations in the nine families with XLOA: five single nucleotide substitutions predicting a change of conserved amino acids (G35D, L39R, D78V, W133R and E233K) when compared with the mouse OA1 homologue, one deletion leading to the skipping of exon 2, and one single nucleotide substitution expected to affect the 5' splice site of intron 2 were found. Subsequent genealogical investigations in the three families harbouring the same mutation disclosed that two of the three pedigrees belonged to the same family. All mutations predict crucial changes in the protein structure. Clinical examination failed to identify any phenotype–genotype pattern except a milder phenotype devoid of iris translucency in the patient with the 5'splice site mutation of intron 2. -

(12) Patent Application Publication (10) Pub. No.: US 2012/025804.6 A1 Mutzke (43) Pub
US 20120258046A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/025804.6 A1 Mutzke (43) Pub. Date: Oct. 11, 2012 (54) MANNOSE-CONTAINING SOLUTION FOR C7H 2L/02 (2006.01) LYOPHILIZATION, TRANSFECTION AND/OR CI2N 15/63 (2006.01) NECTION OF NUCLECACDS A61R 49/00 (2006.01) (76) Inventor: Thorsten Mutzke, Reutlingen (DE) (52) U.S. Cl. ....... 424/9.1: 435/.440; 514/44 R: 514/44 A: 536/23.1:536/24.5:536/24.3: 536/24.33; (21) Appl. No.: 131509,564 424/184.1 (22) PCT Filed: Nov. 8, 2010 (57) ABSTRACT (86). PCT No.: PCT/EP2010/006788 The present invention is directed to (the use of) a solution containing at least one nucleic acid (sequence) and free man S371 (c)(1), nose for lyophilization, transfection and/or injection, particu (2), (4) Date: May 11, 2012 larly of RNA and mRNA. The inventive solution exhibits a positive effect on Stabilization of the nucleic acid (sequence) (30) Foreign Application Priority Data during lyophilization and storage but also leads to a consid erable increase of the transfection efficiency of a nucleic acid. Dec. 9, 2009 (EP) ................... PCTAEP2009/OO8804 It thus also increases in vivo expression of a protein encoded by Such a nucleic acid upon increased transfection rate. The Publication Classification present invention is furthermore directed to a method of lyo (51) Int. Cl. philization using the mannose-containing solution, to phar A6 IK3I/7088 (2006.01) maceutical compositions, vaccines, kits, first and second A6 IK3I/73 (2006.01) medical uses applying such a mannose-containing Solution A6IP37/04 (2006.01) and/or a nucleic acid (sequence) lyophilized or resuspended C7H 2L/04 (2006.01) with Such a solution. -

Download CGT Exome V2.0
CGT Exome version 2. -

Nitisinone for Treatment of Oculocutaneous/Ocular
(19) TZZ ¥¥_T (11) EP 2 538 936 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 31/122 (2006.01) A61P 17/00 (2006.01) 24.12.2014 Bulletin 2014/52 (86) International application number: (21) Application number: 11706137.4 PCT/US2011/026260 (22) Date of filing: 25.02.2011 (87) International publication number: WO 2011/106655 (01.09.2011 Gazette 2011/35) (54) NITISINONE FOR TREATMENT OF OCULOCUTANEOUS/OCULAR ALBINISM AND FOR INCREASING PIGMENTATION NITISINON ZUR BEHANDLUNG VON AUGEN-HAUT-/AUGENALBINISMUS UND FÜR ERHÖHTE PIGMENTIERUNG NITISINONE POUR LE TRAITEMENT DE L’ALBINISME OCULO-CUTANÉ/OCULAIRE ET POUR L’AUGMENTATION DE LA PIGMENTATION (84) Designated Contracting States: (56) References cited: AL AT BE BG CH CY CZ DE DK EE ES FI FR GB • SUZUKI T ET AL: "Recent advances in genetic GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO analysesof oculocutaneous albinismtypes 2 and PL PT RO RS SE SI SK SM TR 4", JOURNAL OF DERMATOLOGICAL SCIENCE, ELSEVIER SCIENCE PUBLISHERS, SHANNON, (30) Priority: 26.02.2010 US 308771 P IE, vol. 51, no. 1, 1 July 2008 (2008-07-01), pages 1-9, XP022638851, ISSN: 0923-1811, DOI: DOI: (43) Date of publication of application: 10.1016/J.JDERMSCI.2007.12.008 [retrieved on 02.01.2013 Bulletin 2013/01 2008-04-14] • SCHIAFFINO MARIA VITTORIA ET AL: "Effective (73) Proprietor: The United States Of America, As retrovirus-mediated gene transfer in normal and Represented By mutant human melanocytes", HUMAN GENE The Secretary, Department Of Health and THERAPY,MARY ANN LIEBERT, NEW YORK , NY, Human Services US, vol. -

(PMEL) Expression Outside of Pigmented Bovine Skin Suggests Functions Beyond Eumelanogenesis
G C A T T A C G G C A T genes Article Indication of Premelanosome Protein (PMEL) Expression Outside of Pigmented Bovine Skin Suggests Functions Beyond Eumelanogenesis Jacqueline Knaust 1, Rosemarie Weikard 1 , Elke Albrecht 2 , Ronald M. Brunner 1, Juliane Günther 1 and Christa Kühn 1,3,* 1 Institute for Genome Biology, Leibniz Institute for Farm Animal Biology (FBN), 18196 Dummerstorf, Germany; [email protected] (J.K.); [email protected] (R.W.); [email protected] (R.M.B.); [email protected] (J.G.) 2 Institute for Muscle Biology and Growth, Leibniz Institute for Farm Animal Biology (FBN), 18196 Dummerstorf, Germany; [email protected] 3 Faculty of Agricultural and Environmental Sciences, University Rostock, 18059 Rostock, Germany * Correspondence: [email protected]; Tel.: +49-38208-68700 Received: 13 May 2020; Accepted: 7 July 2020; Published: 13 July 2020 Abstract: The premelanosome protein (PMEL) is important for fibril formation within melanosomes during vertebrate melanogenesis. Fibrils form a matrix for pigment deposition within pigmented tissues such as skin and hair. PMEL mutations are known to modulate eumelanic pigmentation in vertebrates. However, in bovines, PMEL mutations were also found to alter pheomelanic pigmentation resulting in coat color dilution. Furthermore, epistatic effects of a mutated PMEL allele were detected in the phenotypic expression of the bovine hair defect “rat-tail syndrome” (RTS) characterized by charcoal coat color and hair deformation. Reports about PMEL gene expression in non-pigmented tissues raised the hypothesis that there may be unknown functions of the PMEL protein beyond eumelanin deposition to PMEL fibrils. In our study, we analysed the PMEL protein expression in pigmented skin and non-pigmented bovine tissues (non-pigmented skin, thyroid gland, rumen, liver, kidney, and adrenal gland cortex).