Nitisinone for Treatment of Oculocutaneous/Ocular
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Melanocytes and Their Diseases
Downloaded from http://perspectivesinmedicine.cshlp.org/ on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Melanocytes and Their Diseases Yuji Yamaguchi1 and Vincent J. Hearing2 1Medical, AbbVie GK, Mita, Tokyo 108-6302, Japan 2Laboratory of Cell Biology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892 Correspondence: [email protected] Human melanocytes are distributed not only in the epidermis and in hair follicles but also in mucosa, cochlea (ear), iris (eye), and mesencephalon (brain) among other tissues. Melano- cytes, which are derived from the neural crest, are unique in that they produce eu-/pheo- melanin pigments in unique membrane-bound organelles termed melanosomes, which can be divided into four stages depending on their degree of maturation. Pigmentation production is determined by three distinct elements: enzymes involved in melanin synthesis, proteins required for melanosome structure, and proteins required for their trafficking and distribution. Many genes are involved in regulating pigmentation at various levels, and mutations in many of them cause pigmentary disorders, which can be classified into three types: hyperpigmen- tation (including melasma), hypopigmentation (including oculocutaneous albinism [OCA]), and mixed hyper-/hypopigmentation (including dyschromatosis symmetrica hereditaria). We briefly review vitiligo as a representative of an acquired hypopigmentation disorder. igments that determine human skin colors somes can be divided into four stages depend- Pinclude melanin, hemoglobin (red), hemo- ing on their degree of maturation. Early mela- siderin (brown), carotene (yellow), and bilin nosomes, especially stage I melanosomes, are (yellow). Among those, melanins play key roles similar to lysosomes whereas late melanosomes in determining human skin (and hair) pigmen- contain a structured matrix and highly dense tation. -

EXTENDED CARRIER SCREENING Peace of Mind for Planned Pregnancies
Focusing on Personalised Medicine EXTENDED CARRIER SCREENING Peace of Mind for Planned Pregnancies Extended carrier screening is an important tool for prospective parents to help them determine their risk of having a child affected with a heritable disease. In many cases, parents aren’t aware they are carriers and have no family history due to the rarity of some diseases in the general population. What is covered by the screening? Genomics For Life offers a comprehensive Extended Carrier Screening test, providing prospective parents with the information they require when planning their pregnancy. Extended Carrier Screening has been shown to detect carriers who would not have been considered candidates for traditional risk- based screening. With a simple mouth swab collection, we are able to test for over 419 genes associated with inherited diseases, including Fragile X Syndrome, Cystic Fibrosis and Spinal Muscular Atrophy. The assay has been developed in conjunction with clinical molecular geneticists, and includes genes listed in the NIH Genetic Test Registry. For a list of genes and disorders covered, please see the reverse of this brochure. If your gene of interest is not covered on our Extended Carrier Screening panel, please contact our friendly team to assist you in finding a gene test panel that suits your needs. Why have Extended Carrier Screening? Extended Carrier Screening prior to pregnancy enables couples to learn about their reproductive risk and consider a complete range of reproductive options, including whether or not to become pregnant, whether to use advanced reproductive technologies, such as preimplantation genetic diagnosis, or to use donor gametes. -

Dermatologic Manifestations of Hermansky-Pudlak Syndrome in Patients with and Without a 16–Base Pair Duplication in the HPS1 Gene
STUDY Dermatologic Manifestations of Hermansky-Pudlak Syndrome in Patients With and Without a 16–Base Pair Duplication in the HPS1 Gene Jorge Toro, MD; Maria Turner, MD; William A. Gahl, MD, PhD Background: Hermansky-Pudlak syndrome (HPS) con- without the duplication were non–Puerto Rican except sists of oculocutaneous albinism, a platelet storage pool de- 4 from central Puerto Rico. ficiency, and lysosomal accumulation of ceroid lipofuscin. Patients with HPS from northwest Puerto Rico are homozy- Results: Both patients homozygous for the 16-bp du- gous for a 16–base pair (bp) duplication in exon 15 of HPS1, plication and patients without the duplication dis- a gene on chromosome 10q23 known to cause the disorder. played skin color ranging from white to light brown. Pa- tients with the duplication, as well as those lacking the Objective: To determine the dermatologic findings of duplication, had hair color ranging from white to brown patients with HPS. and eye color ranging from blue to brown. New findings in both groups of patients with HPS were melanocytic Design: Survey of inpatients with HPS by physical ex- nevi with dysplastic features, acanthosis nigricans–like amination. lesions in the axilla and neck, and trichomegaly. Eighty percent of patients with the duplication exhibited fea- Setting: National Institutes of Health Clinical Center, tures of solar damage, including multiple freckles, stel- Bethesda, Md (a tertiary referral hospital). late lentigines, actinic keratoses, and, occasionally, basal cell or squamous cell carcinomas. Only 8% of patients Patients: Sixty-five patients aged 3 to 54 years were di- lacking the 16-bp duplication displayed these findings. -

Preconception Carrier Screening
Focusing on Personalised Medicine PRECONCEPTION CARRIER SCREENING Preconception carrier screening is an important tool for prospective parents to help them determine their risk of having a child affected with a heritable disease. In many cases, parents aren’t aware they are carriers and have no family history due to the rarity of some diseases in the general population. What is covered by the screening? Our Inherited Disease Panel tests over 300 genes associated with more than 700 unique commonly inherited diseases, including common forms of inherited deafness, blindness, heart disease, Parkinson’s disease, immunodeficiency, and various ataxias, anaemias, and treatable metabolic syndromes. The assay has been developed in conjunction with clinical molecular geneticists, and includeds genes listed in the NIH Genetic Test Registry. For a full list of genes and disorders covered, please see the reverse of this brochure. Why have Preconception Carrier Screening? Carrier screening prior to pregnancy enables couples to learn about their reproductive risk and consider a complete range of reproductive options, including whether or not to become pregnant, whether to use advanced reproductive technologies, such as preimplantation genetic diagnosis, or use donor gametes. Screening also allows couples to consider prenatal diagnosis and pregnancy management options in the event of an affected fetus. Whilst individually each disease tested is rare, around 25% of people will carry at least one abnormal mutation. These disorders are usually autosomal recessive, which means that a child must inherit a defective gene from each parent to have the disease. For autosomal recessive conditions, if a person is a carrier of the disease, they have one defective copy of the gene and one normal copy and typically don’t have any symptoms of the disease. -

Ophthalmology
Ophthalmology Information for health professionals MEDICAL GENETIC TESTING FOR OPHTHALMOLOGY Recent technologies, in particularly Next Generation Sequencing (NGS), allows fast, accurate and valuable diagnostic tests. For Ophthalmology, CGC Genetics has an extensive list of medical genetic tests with clinical integration of results by our Medical Geneticists. 1. EXOME SEQUENCING: Exome Sequencing is a very efficient strategy to study most exons of a patient’s genome, unraveling mutations associated with specific disorders or phenotypes. With this diagnostic strategy, patients can be studied with a significantly reduced turnaround time and cost. CGC Genetics has available 2 options for Exome Sequencing: • Whole Exome Sequencing (WES), which analyzes the entire exome (about 20 000 genes); • Disease Exome by CGC Genetics, which analyzes about 6 000 clinically-relevant genes. Any of these can be performed in the index case or in a Trio. 2. NGS PANELS For NGS panels, several genes associated with the same phenotype are simultaneously sequenced. These panels provide increased diagnostic capability with a significantly reduced turnaround time and cost. CGC Genetics has several NGS panels for Ophthalmology that are constantly updated (www.cgcgenetics.com). Any gene studied in exome or NGS panel can also be individually sequenced and analyzed for deletion/duplication events. 3. EXPERTISE IN MEDICAL GENETICS CGC Genetics has Medical Geneticists specialized in genetic counseling for ophthalmological diseases who may advice in choosing the most appropriate -

Oculocutaneous Albinism: an African Perspective
Br Ir Orthopt J 2014; 11: 3–8 Oculocutaneous albinism: an African perspective GERALDINE R. McBRIDE1,2 MSc 1Orthoptic Department, Eye Clinic, University Hospital Galway, Galway, Ireland 2Kwale District Eye Centre, Kwale, Mombasa, Kenya Abstract behind OCA, and explore OCA in an African context in terms of the effects on the health and education of Aim: To describe the genetics behind oculocutaneous individuals with OCA. The outcome of this review will albinism (OCA), and explore OCA in an African be to provide useful advice to those with OCA and those context in terms of the effects on the health and working with or teaching students with OCA. education of individuals with OCA. Methods: A literature-based review was conducted using Pubmed. Searches were restricted to English- Genetics based publications, focusing on OCA in Africa. There are four different types of OCA, which result from Results: The genetics behind OCA and the effects of a mutation in one of several genes (Table 1).1,2,4 Each of OCA in terms of visual impairment and skin cancer these genes is chemically coded to produce proteins are explained along with a description of what low which are involved in the production of melanin. The vision services and low vision aids are available to mutation can result in the reduction of melanin pro- those with OCA in Africa. duction, or no melanin production.1 All four types of Conclusions: The review concludes with useful advice OCA are autosomal recessive. Therefore if both parents to those with OCA and those working with or are carriers of the mutated gene there is a 25% chance of teaching students with OCA. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Oculocutaneous Albinism, a Family Matter Summer Moon, DO,* Katherine Braunlich, DO,** Howard Lipkin, DO,*** Annette Lacasse, DO***
Oculocutaneous Albinism, A Family Matter Summer Moon, DO,* Katherine Braunlich, DO,** Howard Lipkin, DO,*** Annette LaCasse, DO*** *Dermatology Resident, 3rd year, Botsford Hospital Dermatology Residency Program, Farmington Hills, MI **Traditional Rotating Intern, Largo Medical Center, Largo, FL ***Program Director, Botsford Hospital Dermatology Residency Program, Farmington Hills, MI Disclosures: None Correspondence: Katherine Braunlich, DO; [email protected] Abstract Oculocutaneous albinism (OCA) is a group of autosomal-recessive conditions characterized by mutations in melanin biosynthesis with resultant absence or reduction of melanin in the melanocytes. Herein, we present a rare case of two Caucasian sisters diagnosed with oculocutaneous albinism type 1 (OCA1). On physical exam, the sisters had nominal cutaneous evidence of OCA. This case highlights the difficulty of diagnosing oculocutaneous albinism in Caucasians. Additionally, we emphasize the uncommon underlying genetic mutations observed in individuals with oculocutaneous albinism. 2,5 Introduction people has one of the four types of albinism. of exon 4. Additionally, patient A was found to Oculocutaneous albinism (OCA) is a group of We present a rare case of sisters diagnosed with possess the c.21delC frameshift mutation in the autosomal-recessive conditions characterized by oculocutaneous albinism type 1, emphasizing the C10orf11 gene. Patient B was found to possess the mutations in melanin biosynthesis with resultant uncommon genetic mutations we observed in these same heterozygous mutation and deletion in the two individuals. absence or reduction of melanin in the melanocytes. Figure 1 Melanin-poor, pigment-poor melanocytes phenotypically present as hypopigmentation of the Case Report 1,2 Two Caucasian sisters were referred to our hair, skin, and eyes. dermatology clinic after receiving a diagnosis of There are four genes responsible for the four principal oculocutaneous albinism type 1. -

Albinism: Modern Molecular Diagnosis
British Journal of Ophthalmology 1998;82:189–195 189 Br J Ophthalmol: first published as 10.1136/bjo.82.2.189 on 1 February 1998. Downloaded from PERSPECTIVE Albinism: modern molecular diagnosis Susan M Carden, Raymond E Boissy, Pamela J Schoettker, William V Good Albinism is no longer a clinical diagnosis. The past cytes and into which melanin is confined. In the skin, the classification of albinism was predicated on phenotypic melanosome is later transferred from the melanocyte to the expression, but now molecular biology has defined the surrounding keratinocytes. The melanosome precursor condition more accurately. With recent advances in arises from the smooth endoplasmic reticulum. Tyrosinase molecular research, it is possible to diagnose many of the and other enzymes regulating melanin synthesis are various albinism conditions on the basis of genetic produced in the rough endoplasmic reticulum, matured in causation. This article seeks to review the current state of the Golgi apparatus, and translocated to the melanosome knowledge of albinism and associated disorders of hypo- where melanin biosynthesis occurs. pigmentation. Tyrosinase is a copper containing, monophenol, mono- The term albinism (L albus, white) encompasses geneti- oxygenase enzyme that has long been known to have a cally determined diseases that involve a disorder of the critical role in melanogenesis.5 It catalyses three reactions melanin system. Each condition of albinism is due to a in the melanin pathway. The rate limiting step is the genetic mutation on a diVerent chromosome. The cutane- hydroxylation of tyrosine into dihydroxyphenylalanine ous hypopigmentation in albinism ranges from complete (DOPA) by tyrosinase, but tyrosinase does not act alone. -

A Neuroendocrine Theory for the Etiology of Vitiligo
Cush ZM, J Med Stud Res 2018, 1: 007 DOI: 10.24966/MSR-5657/100007 HSOA Journal of Medicine: Study & Research Research Article of the skin, the melanocytes. The triggers, which range from sunburn A Neuroendocrine Theory for to mechanical trauma and chemical exposures, ultimately cause an autoimmune response those targets melanocytes, driving progressive the Etiology of Vitiligo skin depigmentation [3]. Zakiya M Cush* The New York Medical College, Valhalla, New York, USA Figure 1: Vitiligo is a condition in which the skin loses its pigment cells (melanocytes). This can result in discolored patches in different areas of the body, including the skin, hair, retina and mucous membranes [2]. Biochemistry of melanin formation Abstract According to published research, the adenylate cyclase signaling It has long been believed that vitiligo is an autoimmune disease. pathway, the stimulation of protein kinase C via DAG and the ty- And while there are many theories of this disease process, which rosine kinase activity mechanism (Figure 2) are responsible for the include cytotoxic and hereditary, there is another approach, which proliferation of melanocytes in mammals [4]. has yet to be considered. This paper will present a neuroendocrine hypothesis to why vitiligo occurs. With the understanding of the ty- rosinase enzyme, tyrosine kinase activity, and the Melanocytic Stim- ulating Hormone (MSH-α) and its effect on melanocyte production, an alternative approach to vitiligo should be considered, and it be- gins in the brain. Introduction What is vitiligo? Vitiligo is an acquired cutaneous disorder of pigmentation, with an incidence of 0.5% to 2% worldwide which, manifests as white mac- ules on the skin (Figure 1) and can cause significant psychological stress and stigmatization [1,2]. -

Social Discrimination Against People with Albinism
Social discrimination against people with albinism * Mariah Nebre B.A. Candidate, Department of Sociology, California State University Stanislaus, 1 University Circle, Turlock, CA 95382 Received 16 April, 2018; accepted 15 May 2018 Abstract People with albinism have faced different forms of discrimination due to their genetic condition called oculocutaneous albinism. Oculocutaneous albinism, also known as OCA, is a genetic condition that results in people with low skin pigmentation and melanin levels in their hair, skin, and eyes to varying degrees. People who are afflicted with OCA have a high percentage of visual impairment and life- threating sensitivity to the sun. Affected individuals also face negative outcomes of social, cultural, and economic prejudice because of the lack of color that their skin provides. People with albinism are shunned by the rest of their community and are at a high risk of being killed because of their unique physical features. The most common location where people with albinism are discriminated against is in Africa. In this exploratory study, research was conducted by interviewing four people with albinism to get an understanding of the social discrimination they faced. In these in-depth interviews, the focus was on the social aspect of their past and present lives and how they go about addressing any positive and/or negative outcomes they face in society because of their genetic condition. In addition, undergraduate students were surveyed from different fields of study to get an understanding of how much knowledge students have about albinism and its social aspects. Some of the majors surveyed include: biology, sociology, psychology, and liberal studies. -
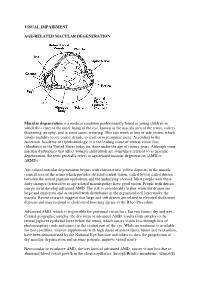
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye.