X-Linked Ocular Albinism: Prevalence and Mutations – a National Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Testing for Non-Cancerous Inheritable Diseases
Genetic Testing for Non-Cancerous Inheritable Diseases Policy Number: Original Effective Date: MM.02.009 03/01/2010 Line(s) of Business: Current Effective Date: HMO; PPO; QUEST Integration 12/30/2016 Section: Medicine Place(s) of Service: Outpatient I. Description A genetic test is the analysis of human DNA, RNA, chromosomes, proteins, or certain metabolites in order to detect alterations related to an inheritable disorder. This can be accomplished by directly examining the DNA or RNA that makes up a gene (direct testing), looking at markers co-inherited with a disease-causing gene (linkage testing), assaying certain metabolites (biochemical testing), or examining the chromosomes (cytogenetic testing). Genetic tests are conducted for a number of purposes, including predicting disease risk, newborn screening, determining clinical management, identifying carriers, and establishing prenatal or clinical diagnoses or prognoses in individuals, families, or populations. Expanded Carrier Screening The American College of Medical Genetics (ACMG ) defines expanded panels as those that use next- generation sequencing to screen for mutations in many genes, as opposed to gene-by-gene screening (e.g., ethnic-specific screening or panethnic testing for cystic fibrosis). An ACMG position statement states that although commercial laboratories offer expanded carrier screening panels, there has been no professional guidance as to which disease genes and mutations to include. This policy does not address oncology-related genetic testing or pre-implantation genetic diagnosis (PGD). II. Criteria/Guidelines Note: For familial assessments, unless otherwise specified, family will be considered: first-, second- and third- degree relatives on the same side of the family. The maternal and paternal sides should be considered independently. -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Preconception Carrier Screening
Focusing on Personalised Medicine PRECONCEPTION CARRIER SCREENING Preconception carrier screening is an important tool for prospective parents to help them determine their risk of having a child affected with a heritable disease. In many cases, parents aren’t aware they are carriers and have no family history due to the rarity of some diseases in the general population. What is covered by the screening? Our Inherited Disease Panel tests over 300 genes associated with more than 700 unique commonly inherited diseases, including common forms of inherited deafness, blindness, heart disease, Parkinson’s disease, immunodeficiency, and various ataxias, anaemias, and treatable metabolic syndromes. The assay has been developed in conjunction with clinical molecular geneticists, and includeds genes listed in the NIH Genetic Test Registry. For a full list of genes and disorders covered, please see the reverse of this brochure. Why have Preconception Carrier Screening? Carrier screening prior to pregnancy enables couples to learn about their reproductive risk and consider a complete range of reproductive options, including whether or not to become pregnant, whether to use advanced reproductive technologies, such as preimplantation genetic diagnosis, or use donor gametes. Screening also allows couples to consider prenatal diagnosis and pregnancy management options in the event of an affected fetus. Whilst individually each disease tested is rare, around 25% of people will carry at least one abnormal mutation. These disorders are usually autosomal recessive, which means that a child must inherit a defective gene from each parent to have the disease. For autosomal recessive conditions, if a person is a carrier of the disease, they have one defective copy of the gene and one normal copy and typically don’t have any symptoms of the disease. -

Ophthalmology
Ophthalmology Information for health professionals MEDICAL GENETIC TESTING FOR OPHTHALMOLOGY Recent technologies, in particularly Next Generation Sequencing (NGS), allows fast, accurate and valuable diagnostic tests. For Ophthalmology, CGC Genetics has an extensive list of medical genetic tests with clinical integration of results by our Medical Geneticists. 1. EXOME SEQUENCING: Exome Sequencing is a very efficient strategy to study most exons of a patient’s genome, unraveling mutations associated with specific disorders or phenotypes. With this diagnostic strategy, patients can be studied with a significantly reduced turnaround time and cost. CGC Genetics has available 2 options for Exome Sequencing: • Whole Exome Sequencing (WES), which analyzes the entire exome (about 20 000 genes); • Disease Exome by CGC Genetics, which analyzes about 6 000 clinically-relevant genes. Any of these can be performed in the index case or in a Trio. 2. NGS PANELS For NGS panels, several genes associated with the same phenotype are simultaneously sequenced. These panels provide increased diagnostic capability with a significantly reduced turnaround time and cost. CGC Genetics has several NGS panels for Ophthalmology that are constantly updated (www.cgcgenetics.com). Any gene studied in exome or NGS panel can also be individually sequenced and analyzed for deletion/duplication events. 3. EXPERTISE IN MEDICAL GENETICS CGC Genetics has Medical Geneticists specialized in genetic counseling for ophthalmological diseases who may advice in choosing the most appropriate -

Diseases of the Digestive System (KOO-K93)
CHAPTER XI Diseases of the digestive system (KOO-K93) Diseases of oral cavity, salivary glands and jaws (KOO-K14) lijell Diseases of pulp and periapical tissues 1m Dentofacial anomalies [including malocclusion] Excludes: hemifacial atrophy or hypertrophy (Q67.4) K07 .0 Major anomalies of jaw size Hyperplasia, hypoplasia: • mandibular • maxillary Macrognathism (mandibular)(maxillary) Micrognathism (mandibular)( maxillary) Excludes: acromegaly (E22.0) Robin's syndrome (087.07) K07 .1 Anomalies of jaw-cranial base relationship Asymmetry of jaw Prognathism (mandibular)( maxillary) Retrognathism (mandibular)(maxillary) K07.2 Anomalies of dental arch relationship Cross bite (anterior)(posterior) Dis to-occlusion Mesio-occlusion Midline deviation of dental arch Openbite (anterior )(posterior) Overbite (excessive): • deep • horizontal • vertical Overjet Posterior lingual occlusion of mandibular teeth 289 ICO-N A K07.3 Anomalies of tooth position Crowding Diastema Displacement of tooth or teeth Rotation Spacing, abnormal Transposition Impacted or embedded teeth with abnormal position of such teeth or adjacent teeth K07.4 Malocclusion, unspecified K07.5 Dentofacial functional abnormalities Abnormal jaw closure Malocclusion due to: • abnormal swallowing • mouth breathing • tongue, lip or finger habits K07.6 Temporomandibular joint disorders Costen's complex or syndrome Derangement of temporomandibular joint Snapping jaw Temporomandibular joint-pain-dysfunction syndrome Excludes: current temporomandibular joint: • dislocation (S03.0) • strain (S03.4) K07.8 Other dentofacial anomalies K07.9 Dentofacial anomaly, unspecified 1m Stomatitis and related lesions K12.0 Recurrent oral aphthae Aphthous stomatitis (major)(minor) Bednar's aphthae Periadenitis mucosa necrotica recurrens Recurrent aphthous ulcer Stomatitis herpetiformis 290 DISEASES OF THE DIGESTIVE SYSTEM Diseases of oesophagus, stomach and duodenum (K20-K31) Ill Oesophagitis Abscess of oesophagus Oesophagitis: • NOS • chemical • peptic Use additional external cause code (Chapter XX), if desired, to identify cause. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Oculocutaneous Albinism, a Family Matter Summer Moon, DO,* Katherine Braunlich, DO,** Howard Lipkin, DO,*** Annette Lacasse, DO***
Oculocutaneous Albinism, A Family Matter Summer Moon, DO,* Katherine Braunlich, DO,** Howard Lipkin, DO,*** Annette LaCasse, DO*** *Dermatology Resident, 3rd year, Botsford Hospital Dermatology Residency Program, Farmington Hills, MI **Traditional Rotating Intern, Largo Medical Center, Largo, FL ***Program Director, Botsford Hospital Dermatology Residency Program, Farmington Hills, MI Disclosures: None Correspondence: Katherine Braunlich, DO; [email protected] Abstract Oculocutaneous albinism (OCA) is a group of autosomal-recessive conditions characterized by mutations in melanin biosynthesis with resultant absence or reduction of melanin in the melanocytes. Herein, we present a rare case of two Caucasian sisters diagnosed with oculocutaneous albinism type 1 (OCA1). On physical exam, the sisters had nominal cutaneous evidence of OCA. This case highlights the difficulty of diagnosing oculocutaneous albinism in Caucasians. Additionally, we emphasize the uncommon underlying genetic mutations observed in individuals with oculocutaneous albinism. 2,5 Introduction people has one of the four types of albinism. of exon 4. Additionally, patient A was found to Oculocutaneous albinism (OCA) is a group of We present a rare case of sisters diagnosed with possess the c.21delC frameshift mutation in the autosomal-recessive conditions characterized by oculocutaneous albinism type 1, emphasizing the C10orf11 gene. Patient B was found to possess the mutations in melanin biosynthesis with resultant uncommon genetic mutations we observed in these same heterozygous mutation and deletion in the two individuals. absence or reduction of melanin in the melanocytes. Figure 1 Melanin-poor, pigment-poor melanocytes phenotypically present as hypopigmentation of the Case Report 1,2 Two Caucasian sisters were referred to our hair, skin, and eyes. dermatology clinic after receiving a diagnosis of There are four genes responsible for the four principal oculocutaneous albinism type 1. -

Albinism: Modern Molecular Diagnosis
British Journal of Ophthalmology 1998;82:189–195 189 Br J Ophthalmol: first published as 10.1136/bjo.82.2.189 on 1 February 1998. Downloaded from PERSPECTIVE Albinism: modern molecular diagnosis Susan M Carden, Raymond E Boissy, Pamela J Schoettker, William V Good Albinism is no longer a clinical diagnosis. The past cytes and into which melanin is confined. In the skin, the classification of albinism was predicated on phenotypic melanosome is later transferred from the melanocyte to the expression, but now molecular biology has defined the surrounding keratinocytes. The melanosome precursor condition more accurately. With recent advances in arises from the smooth endoplasmic reticulum. Tyrosinase molecular research, it is possible to diagnose many of the and other enzymes regulating melanin synthesis are various albinism conditions on the basis of genetic produced in the rough endoplasmic reticulum, matured in causation. This article seeks to review the current state of the Golgi apparatus, and translocated to the melanosome knowledge of albinism and associated disorders of hypo- where melanin biosynthesis occurs. pigmentation. Tyrosinase is a copper containing, monophenol, mono- The term albinism (L albus, white) encompasses geneti- oxygenase enzyme that has long been known to have a cally determined diseases that involve a disorder of the critical role in melanogenesis.5 It catalyses three reactions melanin system. Each condition of albinism is due to a in the melanin pathway. The rate limiting step is the genetic mutation on a diVerent chromosome. The cutane- hydroxylation of tyrosine into dihydroxyphenylalanine ous hypopigmentation in albinism ranges from complete (DOPA) by tyrosinase, but tyrosinase does not act alone. -

Soonerstart Automatic Qualifying Syndromes and Conditions
SoonerStart Automatic Qualifying Syndromes and Conditions - Appendix O Abetalipoproteinemia Acanthocytosis (see Abetalipoproteinemia) Accutane, Fetal Effects of (see Fetal Retinoid Syndrome) Acidemia, 2-Oxoglutaric Acidemia, Glutaric I Acidemia, Isovaleric Acidemia, Methylmalonic Acidemia, Propionic Aciduria, 3-Methylglutaconic Type II Aciduria, Argininosuccinic Acoustic-Cervico-Oculo Syndrome (see Cervico-Oculo-Acoustic Syndrome) Acrocephalopolysyndactyly Type II Acrocephalosyndactyly Type I Acrodysostosis Acrofacial Dysostosis, Nager Type Adams-Oliver Syndrome (see Limb and Scalp Defects, Adams-Oliver Type) Adrenoleukodystrophy, Neonatal (see Cerebro-Hepato-Renal Syndrome) Aglossia Congenita (see Hypoglossia-Hypodactylia) Aicardi Syndrome AIDS Infection (see Fetal Acquired Immune Deficiency Syndrome) Alaninuria (see Pyruvate Dehydrogenase Deficiency) Albers-Schonberg Disease (see Osteopetrosis, Malignant Recessive) Albinism, Ocular (includes Autosomal Recessive Type) Albinism, Oculocutaneous, Brown Type (Type IV) Albinism, Oculocutaneous, Tyrosinase Negative (Type IA) Albinism, Oculocutaneous, Tyrosinase Positive (Type II) Albinism, Oculocutaneous, Yellow Mutant (Type IB) Albinism-Black Locks-Deafness Albright Hereditary Osteodystrophy (see Parathyroid Hormone Resistance) Alexander Disease Alopecia - Mental Retardation Alpers Disease Alpha 1,4 - Glucosidase Deficiency (see Glycogenosis, Type IIA) Alpha-L-Fucosidase Deficiency (see Fucosidosis) Alport Syndrome (see Nephritis-Deafness, Hereditary Type) Amaurosis (see Blindness) Amaurosis -

Hereditary Hearing Impairment with Cutaneous Abnormalities
G C A T T A C G G C A T genes Review Hereditary Hearing Impairment with Cutaneous Abnormalities Tung-Lin Lee 1 , Pei-Hsuan Lin 2,3, Pei-Lung Chen 3,4,5,6 , Jin-Bon Hong 4,7,* and Chen-Chi Wu 2,3,5,8,* 1 Department of Medical Education, National Taiwan University Hospital, Taipei City 100, Taiwan; [email protected] 2 Department of Otolaryngology, National Taiwan University Hospital, Taipei 11556, Taiwan; [email protected] 3 Graduate Institute of Clinical Medicine, National Taiwan University College of Medicine, Taipei City 100, Taiwan; [email protected] 4 Graduate Institute of Medical Genomics and Proteomics, National Taiwan University College of Medicine, Taipei City 100, Taiwan 5 Department of Medical Genetics, National Taiwan University Hospital, Taipei 10041, Taiwan 6 Department of Internal Medicine, National Taiwan University Hospital, Taipei 10041, Taiwan 7 Department of Dermatology, National Taiwan University Hospital, Taipei City 100, Taiwan 8 Department of Medical Research, National Taiwan University Biomedical Park Hospital, Hsinchu City 300, Taiwan * Correspondence: [email protected] (J.-B.H.); [email protected] (C.-C.W.) Abstract: Syndromic hereditary hearing impairment (HHI) is a clinically and etiologically diverse condition that has a profound influence on affected individuals and their families. As cutaneous findings are more apparent than hearing-related symptoms to clinicians and, more importantly, to caregivers of affected infants and young individuals, establishing a correlation map of skin manifestations and their underlying genetic causes is key to early identification and diagnosis of syndromic HHI. In this article, we performed a comprehensive PubMed database search on syndromic HHI with cutaneous abnormalities, and reviewed a total of 260 relevant publications. -

Analysis of Ocular Hypopigmentation in Rab38cht/Cht Mice
ARTICLES Analysis of Ocular Hypopigmentation in Rab38cht/cht Mice Brian P. Brooks,1,2,3 Denise M. Larson,2,3 Chi-Chao Chan,1 Sten Kjellstrom,4 Richard S. Smith,5,6 Mary A. Crawford,1 Lynn Lamoreux,7 Marjan Huizing,3 Richard Hess,3 Xiaodong Jiao,1 J. Fielding Hejtmancik,1 Arvydas Maminishkis,1 Simon W. M. John,5,6 Ronald Bush,4 and William J. Pavan3 cht PURPOSE. To characterize the ocular phenotype resulting from and RPE thinning. The synergistic effects of the Rab38 and mutation of Rab38, a candidate gene for Hermansky-Pudlak Tyrp1b alleles suggest that TYRP1 is not the only target of syndrome. RAB38 trafficking. This mouse line provides a useful model for cht/cht METHODS. Chocolate mice (cht, Rab38 ) and control het- studying melanosome biology and its role in human ocular erozygous (Rab38cht/ϩ) and wild-type mice were examined diseases. (Invest Ophthalmol Vis Sci. 2007;48:3905–3913) clinically, histologically, ultrastructurally, and electrophysi- DOI:10.1167/iovs.06-1464 ologically. Mice homozygous for both the Rab38cht and the Tyrp1b alleles were similarly examined. he analysis of mice that exhibit defects in coat coloration cht/cht RESULTS. Rab38 mice showed variable peripheral iris trans- T(coat color mutants) has aided in the identification of 1 illumination defects at 2 months of age. Patches of RPE hypo- genes important in eye, skin, and hair pigmentation. Many of pigmentation were noted clinically in 57% of Rab38cht/cht eyes these genes are mutated in patients with pigmentary anoma- and 6% of Rab38cht/ϩ eyes. Rab38cht/cht mice exhibited thin- lies. -
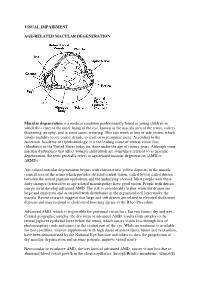
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye.