The Mysterious Orphans of Mycoplasmataceae Tatiana V
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Movements of Mycoplasma Mobile Gliding Machinery Detected by High
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.28.428740; this version posted January 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 mBio (Research Article) 2 3 Movements of Mycoplasma mobile gliding machinery detected by 4 high-speed atomic force microscopy 5 Kohei Kobayashia*, Noriyuki Koderab*, Taishi Kasaia, Yuhei O Taharaa,c, Takuma 6 Toyonagaa, Masaki Mizutania, Ikuko Fujiwaraa, Toshio Andob, Makoto Miyataa,c,# 7 8 aGraduate School of Science, Osaka City University, 3-3-138 Sugimoto, 9 Sumiyoshi-ku, Osaka 558-8585, Japan. 10 bNano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kakuma-chou, 11 Kanazawa, Ishikawa 920-1192, Japan. 12 cThe OCU Advanced Research Institute for Natural Science and Technology 13 (OCARINA), Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 14 558-8585, Japan. 15 16 Address correspondence to Makoto Miyata, [email protected] 17 *These authors contributed equally to this work. 18 Present address: Taishi Kasai: Department of Life Science, Rikkyo University, 19 3-34-1 Nishiikebukuro, Toshima-ku, Tokyo 171-8501, Japan. 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.28.428740; this version posted January 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. -

Simultaneous Identification of Host, Ectoparasite and Pathogen DNA Via
Page 1 of 42 Molecular Ecology Resources 1 Simultaneous identification of host, ectoparasite and 2 pathogen DNA via in-solution capture 3 4 Michael G. Campana* ,1 , Melissa T. R. Hawkins* ,1,2 , Lauren H. Henson 1, Kristin 5 Stewardson 1, Hillary S. Young 3, Leah R. Card 4, Justin Lock 1, Bernard 6 Agwanda 5, Jory Brinkerhoff 6, Holly D. Gaff 7, Kristofer M. Helgen 2, Jesús E. 1, 2 4 1,§ 7 Maldonado , William J. McShea , Robert C. Fleischer 8 *Co-first Authors; §Corresponding author: [email protected] 9 1Center for Conservation and Evolutionary Genetics, Smithsonian Conservation Biology Institute, 10 3001 Connecticut Avenue NW, Washington, DC 20008, USA 11 2Division of Mammals, National Museum of Natural History, MRC 108, Smithsonian Institution, P.O. 12 Box 37012, Washington, DC 20013-7012, USA 13 3Department of Ecology, Evolution and Marine Biology, University of California Santa Barbara, Santa 14 Barbara, CA 93106, USA 15 4Smithsonian Conservation Biology Institute, National Zoological Park, 1500 Remount Rd., Front 16 Royal, VA 22630, USA 17 5National Museums of Kenya, Box 40658-00100, Nairobi, Kenya 18 6Department of Biology, University of Richmond, B322 Gottwald Center for the Sciences, 28 19 Westhampton Way, University of Richmond, VA 23173, USA 20 7Department of Biological Sciences, Old Dominion University, Norfolk, VA 23529, USA 21 22 Keywords: Ectoparasite, blood meal, pathogen, in-solution capture, DNA, high-throughput 23 sequencing 24 Running Title: Host, ectoparasite and pathogen capture 1 Molecular Ecology Resources Page 2 of 42 25 Abstract 26 Ectoparasites frequently vector pathogens from often unknown pathogen reservoirs to 27 both human and animal populations. -

The Mysterious Orphans of Mycoplasmataceae
The mysterious orphans of Mycoplasmataceae Tatiana V. Tatarinova1,2*, Inna Lysnyansky3, Yuri V. Nikolsky4,5,6, and Alexander Bolshoy7* 1 Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, 90027, California, USA 2 Spatial Science Institute, University of Southern California, Los Angeles, 90089, California, USA 3 Mycoplasma Unit, Division of Avian and Aquatic Diseases, Kimron Veterinary Institute, POB 12, Beit Dagan, 50250, Israel 4 School of Systems Biology, George Mason University, 10900 University Blvd, MSN 5B3, Manassas, VA 20110, USA 5 Biomedical Cluster, Skolkovo Foundation, 4 Lugovaya str., Skolkovo Innovation Centre, Mozhajskij region, Moscow, 143026, Russian Federation 6 Vavilov Institute of General Genetics, Moscow, Russian Federation 7 Department of Evolutionary and Environmental Biology and Institute of Evolution, University of Haifa, Israel 1,2 [email protected] 3 [email protected] 4-6 [email protected] 7 [email protected] 1 Abstract Background: The length of a protein sequence is largely determined by its function, i.e. each functional group is associated with an optimal size. However, comparative genomics revealed that proteins’ length may be affected by additional factors. In 2002 it was shown that in bacterium Escherichia coli and the archaeon Archaeoglobus fulgidus, protein sequences with no homologs are, on average, shorter than those with homologs [1]. Most experts now agree that the length distributions are distinctly different between protein sequences with and without homologs in bacterial and archaeal genomes. In this study, we examine this postulate by a comprehensive analysis of all annotated prokaryotic genomes and focusing on certain exceptions. -

MIB–MIP Is a Mycoplasma System That Captures and Cleaves Immunoglobulin G
MIB–MIP is a mycoplasma system that captures and cleaves immunoglobulin G Yonathan Arfia,b,1, Laetitia Minderc,d, Carmelo Di Primoe,f,g, Aline Le Royh,i,j, Christine Ebelh,i,j, Laurent Coquetk, Stephane Claveroll, Sanjay Vasheem, Joerg Joresn,o, Alain Blancharda,b, and Pascal Sirand-Pugneta,b aINRA (Institut National de la Recherche Agronomique), UMR 1332 Biologie du Fruit et Pathologie, F-33882 Villenave d’Ornon, France; bUniversity of Bordeaux, UMR 1332 Biologie du Fruit et Pathologie, F-33882 Villenave d’Ornon, France; cInstitut Européen de Chimie et Biologie, UMS 3033, University of Bordeaux, 33607 Pessac, France; dInstitut Bergonié, SIRIC BRIO, 33076 Bordeaux, France; eINSERM U1212, ARN Regulation Naturelle et Artificielle, 33607 Pessac, France; fCNRS UMR 5320, ARN Regulation Naturelle et Artificielle, 33607 Pessac, France; gInstitut Européen de Chimie et Biologie, University of Bordeaux, 33607 Pessac, France; hInstitut de Biologie Structurale, University of Grenoble Alpes, F-38044 Grenoble, France; iCNRS, Institut de Biologie Structurale, F-38044 Grenoble, France; jCEA, Institut de Biologie Structurale, F-38044 Grenoble, France; kCNRS UMR 6270, Plateforme PISSARO, Institute for Research and Innovation in Biomedicine - Normandie Rouen, Normandie Université, F-76821 Mont-Saint-Aignan, France; lProteome Platform, Functional Genomic Center of Bordeaux, University of Bordeaux, F-33076 Bordeaux Cedex, France; mJ. Craig Venter Institute, Rockville, MD 20850; nInternational Livestock Research Institute, 00100 Nairobi, Kenya; and oInstitute of Veterinary Bacteriology, University of Bern, CH-3001 Bern, Switzerland Edited by Roy Curtiss III, University of Florida, Gainesville, FL, and approved March 30, 2016 (received for review January 12, 2016) Mycoplasmas are “minimal” bacteria able to infect humans, wildlife, introduced into naive herds (8). -

Role of Protein Phosphorylation in Mycoplasma Pneumoniae
Pathogenicity of a minimal organism: Role of protein phosphorylation in Mycoplasma pneumoniae Dissertation zur Erlangung des mathematisch-naturwissenschaftlichen Doktorgrades „Doctor rerum naturalium“ der Georg-August-Universität Göttingen vorgelegt von Sebastian Schmidl aus Bad Hersfeld Göttingen 2010 Mitglieder des Betreuungsausschusses: Referent: Prof. Dr. Jörg Stülke Koreferent: PD Dr. Michael Hoppert Tag der mündlichen Prüfung: 02.11.2010 “Everything should be made as simple as possible, but not simpler.” (Albert Einstein) Danksagung Zunächst möchte ich mich bei Prof. Dr. Jörg Stülke für die Ermöglichung dieser Doktorarbeit bedanken. Nicht zuletzt durch seine freundliche und engagierte Betreuung hat mir die Zeit viel Freude bereitet. Des Weiteren hat er mir alle Freiheiten zur Verwirklichung meiner eigenen Ideen gelassen, was ich sehr zu schätzen weiß. Für die Übernahme des Korreferates danke ich PD Dr. Michael Hoppert sowie Prof. Dr. Heinz Neumann, PD Dr. Boris Görke, PD Dr. Rolf Daniel und Prof. Dr. Botho Bowien für das Mitwirken im Thesis-Komitee. Der Studienstiftung des deutschen Volkes gilt ein besonderer Dank für die finanzielle Unterstützung dieser Arbeit, durch die es mir unter anderem auch möglich war, an Tagungen in fernen Ländern teilzunehmen. Prof. Dr. Michael Hecker und der Gruppe von Dr. Dörte Becher (Universität Greifswald) danke ich für die freundliche Zusammenarbeit bei der Durchführung von zahlreichen Proteomics-Experimenten. Ein ganz besonderer Dank geht dabei an Katrin Gronau, die mich in die Feinheiten der 2D-Gelelektrophorese eingeführt hat. Außerdem möchte ich mich bei Andreas Otto für die zahlreichen Proteinidentifikationen in den letzten Monaten bedanken. Nicht zu vergessen ist auch meine zweite Außenstelle an der Universität in Barcelona. Dr. Maria Lluch-Senar und Dr. -

Correlation Between the Oral Microbiome and Brain Resting State Connectivity in Smokers
bioRxiv preprint doi: https://doi.org/10.1101/444612; this version posted October 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Correlation between the oral microbiome and brain resting state connectivity in smokers Dongdong Lin1, Kent Hutchison2, Salvador Portillo3, Victor Vegara1, Jarod Ellingson2, Jingyu Liu1,3, Amanda Carroll-Portillo3,* ,Vince D. Calhoun1,3,* 1The Mind Research Network, Albuquerque, New Mexico, 87106 2University of Colorado Boulder, Boulder, CO 3University of New Mexico, Department of Electrical and Computer Engineering, Albuquerque, New Mexico, 87106 * authors contributed equally to the work. Abstract Recent studies have shown a critical role for the gastrointestinal microbiome in brain and behavior via a complex gut–microbiome–brain axis, however, the influence of the oral microbiome in neurological processes is much less studied, especially in response to the stimuli in the oral microenvironment such as smoking. Additionally, given the complex structural and functional networks in brain system, our knowledge about the relationship between microbiome and brain functions on specific brain circuits is still very limited. In this pilot work, we leverage next generation microbial sequencing with functional MRI techniques to enable the delineation of microbiome-brain network links as well as their relations to cigarette smoking. Thirty smokers and 30 age- and sex- matched non-smokers were recruited for measuring both microbial community and brain functional networks. Statistical analyses were performed to demonstrate the influence of smoking on: the taxonomy and abundance of the constituents within the oral microbial community, brain functional network connectivity, and associations between microbial shifts and the brain signaling network. -

Genes Involved in Cell Division in Mycoplasmas
Genetics and Molecular Biology, 30, 1, 174-181 (2007) Copyright by the Brazilian Society of Genetics. Printed in Brazil www.sbg.org.br Research Article Genes involved in cell division in mycoplasmas Frank Alarcón1, Ana Tereza Ribeiro de Vasconcelos1, Lucia Yim2 and Arnaldo Zaha3 1Laboratório Nacional de Computação Científica / Ministério da Ciência e Tecnologia, Petrópolis, RJ, Brazil. 2Instituto de Biologia Molecular do Paraná, Curitiba, PR, Brazil. 3Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil. Abstract Bacterial cell division has been studied mainly in model systems such as Escherichia coli and Bacillus subtilis, where it is described as a complex process with the participation of a group of proteins which assemble into a multiprotein complex called the septal ring. Mycoplasmas are cell wall-less bacteria presenting a reduced genome. Thus, it was important to compare their genomes to analyze putative genes involved in cell division processes. The division and cell wall (dcw) cluster, which in E. coli and B. subtilis is composed of 16 and 17 genes, respectively, is represented by only three to four genes in mycoplasmas. Even the most conserved protein, FtsZ, is not present in all mycoplasma genomes analyzed so far. A model for the FtsZ protein from Mycoplasma hyopneumoniae and Mycoplasma synoviae has been constructed. The conserved residues, essential for GTP/GDP binding, are present in FtsZ from both species. A strong conservation of hydrophobic amino acid patterns is observed, and is probably necessary for the structural stability of the protein when active. M. synoviae FtsZ presents an extended amino acid sequence at the C-terminal portion of the protein, which may participate in interactions with other still unknown proteins crucial for the cell division process. -

Mycoplasma Pneumoniae Terminal Organelle
MYCOPLASMA PNEUMONIAE TERMINAL ORGANELLE DEVELOPMENT AND GLIDING MOTILITY by BENJAMIN MICHAEL HASSELBRING (Under the Direction of Duncan Charles Krause) ABSTRACT With a minimal genome containing less than 700 open reading frames and a cell volume < 10% of that of model prokaryotes, Mycoplasma pneumoniae is considered among the smallest and simplest organisms capable of self-replication. And yet, this unique wall-less bacterium exhibits a remarkable level of cellular complexity with a dynamic cytoskeleton and a morphological asymmetry highlighted by a polar, membrane-bound terminal organelle containing an elaborate macromolecular core. The M. pneumoniae terminal organelle functions in distinct, and seemingly disparate cellular processes that include cytadherence, cell division, and presumably gliding motility, as individual cells translocate over surfaces with the cell pole harboring the structure engaged as the leading end. While recent years have witnessed a dramatic increase in the knowledge of protein interactions required for core stability and adhesin trafficking, the mechanism of M. pneumoniae gliding has not been defined nor have interdependencies between the various terminal organelle functions been assessed. The studies presented in the current volume describe the first genetic and molecular investigations into the location, components, architecture, and regulation of the M. pneumoniae gliding machinery. The data indicate that cytadherence and gliding motility are separable properties, and identify a subset of M. pneumoniae proteins contributing directly to the latter process. Characterizations of novel gliding-deficient mutants confirm that the terminal organelle contains the molecular gliding machinery, revealing that with the loss of a single terminal organelle cytoskeletal element, protein P41, terminal organelles detach from the cell body but retain gliding function. -

Structure of Mycoplasma Mobile
Cytoskeletal ‘‘jellyfish’’ structure of Mycoplasma mobile Daisuke Nakane* and Makoto Miyata*†‡ *Graduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka 558-8585, Japan; and †PRESTO, Japan Science and Technology Agency, Sumiyoshi-ku, Osaka 558-8585, Japan Edited by David J. DeRosier, Brandeis University, Waltham, MA, and approved October 16, 2007 (received for review May 8, 2007) Mycoplasma mobile, a parasitic bacterium lacking a peptidoglycan This scenario leads to a crucial question: What physical layer, glides on solid surfaces in the direction of a membrane structure could support a gliding force as strong as 27 pN at protrusion at a cell pole by a unique mechanism. Recently, we maximum, while maintaining the flask cell shape? As in the case proposed a working model in which cells are propelled by leg of other mycoplasmas, M. mobile does not have a bacterial cell proteins clustering at the protrusion’s base. The legs repeatedly wall—i.e., a peptidoglycan layer. Moreover, the genome does not catch and release sialic acids on the solid surface, a motion that is have bacterial cytoskeletal proteins, such as MreB or FtsZ (28, driven by the force generated by ATP hydrolysis. Here, to clarify the 29). M. pneumonia, which is positioned at some distance from M. subcellular structure supporting the gliding force and the cell mobile on the phylogenetic tree in mycoplasmas, also can glide shape, we stripped the membrane by Triton X-100 and identified by its membrane protrusion (3, 4, 10, 30, 46). This species has a a unique structure, designated the ‘‘jellyfish’’ structure. In this cytoskeletal structure in the membrane protrusion, and some of structure, an oval solid ‘‘bell’’ Ϸ235 wide and 155 nm long is filled its protein components have been identified (2–5, 31, 32, 46). -
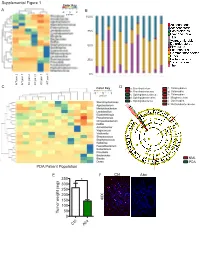
Mental Figure 1 Color Key a -2 0 2 B Z-Score 100%
Supplemental Figure 1 Color Key A -2 0 2 B z-score 100% 75% 50% 25% 0% KC pan 1 WT pan 3 WT KC pan 3 WT pan 2 WT pan 1 WT KC pan 2 C Color Key D a: Brevibacterium f: Chlamydiales b: Brevibacteriaceae g: Chlamydiia -3 0 3 z-score c: Sphingobacteriaceae h: Chlamydiae d: Sphingobacteriales i: Mogibacterium e: Sphingobacteriia j: Oscillospira k: Methylobacteriaceae NML Control Microb.-entrained MΦ PDA PDA Patient Population Control Microb.-entrained MΦ + Myd88i E F Ctrl Abx 350 * 300 250 200 150 40X 100 Tumor weight (mg) 50 0 x Ctrl Ab Supplemental Figure 2 A KC WT B ** * Actinobacteria * ** Bacteroidetes Cyanobacteria Deferribacteres * Firmicutes Proteobacteria % Relative abundance TM7 Others Time(wks) 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 Alpha Diversity Measure C E 60 KC WT 40 20 B. pseudolongum B. animalis 60 5 KC WT 0 B. adolescentis 40% Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 20 Age (weeks) B. pseudolongum B. animalis 5 0 B. adolescentis % Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 F Age (weeks) Week 3 Week 9 Week 13 p=0.678 p=0.02 p=0.385 Time(wks) 3 9 24 20 13 16 D 28 32 36 Week 13 KC WT Firmicutes; Ruminococcus Firmicutes; Dehalobacterium Alpha Diversity Measure Firmicutes; Oscillospira Bacteroidates; Odoribacter Axis.2 [12.7%] Actinobacteria; Bifidobacterium Axis.2 [23.8%] Axis.2 [24.7%] Week 16 Bacteroidetes; Bacteroidales Axis.1 [80.8%] Axis.1 [65.4%] Axis.1 [49.6%] Actinobacteria; Bifidobacterium Week 16 Week 20 Week 24 Week 20 p=0.339 p=0.036 p=0.021 Firmicutes; Dehalobacterium -

Exploring the Cockatiel (Nymphicus Hollandicus) Fecal Microbiome, Bacterial Inhabitants of a Worldwide Pet
Exploring the cockatiel (Nymphicus hollandicus) fecal microbiome, bacterial inhabitants of a worldwide pet Luis David Alcaraz1, Apolinar M. Hernández2 and Mariana Peimbert2 1 Laboratorio Nacional de Ciencias de la Sostenibilidad, Instituto de Ecología, Universidad Nacional Autonóma de México, Mexico City, Mexico 2 Departamento de Ciencias Naturales, Unidad Cuajimalpa, Universidad Autónoma Metropolitana, Mexico City, Mexico ABSTRACT Background. Cockatiels (Nymphicus hollandicus) were originally endemic to Australia; now they are popular pets with a global distribution. It is now possible to conduct detailed molecular studies on cultivable and uncultivable bacteria that are part of the intestinal microbiome of healthy animals. These studies show that bacteria are an essential part of the metabolic capacity of animals. There are few studies on bird microbiomes and, to the best of our knowledge, this is the first report on the cockatiel microbiome. Methods. In this paper, we analyzed the gut microbiome from fecal samples of three healthy adult cockatiels by massive sequencing of the 16S rRNA gene. Additionally, we compared the cockatiel fecal microbiomes with those of other bird species, including poultry and wild birds. Results. The vast majority of the bacteria found in cockatiels were Firmicutes, while Proteobacteria and Bacteroidetes were poorly represented. A total of 19,280 different OTUs were detected, of which 8,072 belonged to the Erysipelotrichaceae family. Discussion. It is relevant to study cockatiel the microbiomes of cockatiels owing to their wide geographic distribution and close human contact. This study serves as a reference for cockatiel bacterial diversity. Despite the large OTU numbers, the diversity is not even Submitted 14 July 2016 Accepted 28 November 2016 and is dominated by Firmicutes of the Erysipelotrichaceae family. -

1 Supplementary Material a Major Clade of Prokaryotes with Ancient
Supplementary Material A major clade of prokaryotes with ancient adaptations to life on land Fabia U. Battistuzzi and S. Blair Hedges Data assembly and phylogenetic analyses Protein data set: Amino acid sequences of 25 protein-coding genes (“proteins”) were concatenated in an alignment of 18,586 amino acid sites and 283 species. These proteins included: 15 ribosomal proteins (RPL1, 2, 3, 5, 6, 11, 13, 16; RPS2, 3, 4, 5, 7, 9, 11), four genes (RNA polymerase alpha, beta, and gamma subunits, Transcription antitermination factor NusG) from the functional category of Transcription, three proteins (Elongation factor G, Elongation factor Tu, Translation initiation factor IF2) of the Translation, Ribosomal Structure and Biogenesis functional category, one protein (DNA polymerase III, beta subunit) of the DNA Replication, Recombination and repair category, one protein (Preprotein translocase SecY) of the Cell Motility and Secretion category, and one protein (O-sialoglycoprotein endopeptidase) of the Posttranslational Modification, Protein Turnover, Chaperones category, as annotated in the Cluster of Orthologous Groups (COG) (Tatusov et al. 2001). After removal of multiple strains of the same species, GBlocks 0.91b (Castresana 2000) was applied to each protein in the concatenation to delete poorly aligned sites (i.e., sites with gaps in more than 50% of the species and conserved in less than 50% of the species) with the following parameters: minimum number of sequences for a conserved position: 110, minimum number of sequences for a flank position: 110, maximum number of contiguous non-conserved positions: 32000, allowed gap positions: with half. The signal-to-noise ratio was determined by altering the “minimum length of a block” parameter.