APCC6 (The 6Th Asia-Pacific Chromosome Colloquium)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2019 Abstrack Book
Table of Contents St. Vincent’s Hospital Map ................................................................................................ 2 Message from the Co-chairs.............................................................................................. 3 Biomed Link 2019 Organising Committee .......................................................................... 4 Biomed Link 2019 Sponsors .............................................................................................. 5 Biomed Link 2019 Session Times ..................................................................................... 10 Welcome Address ........................................................................................................... 12 Keynote Address ............................................................................................................ 13 Biomed Link 2019 3MT Session ....................................................................................... 15 Oral Presentation Abstracts ............................................................................................ 17 Poster Presentation Abstracts ........................................................................................ 33 Judges and Reviewers ................................................................................................... 108 Acknowledgements ...................................................................................................... 109 1 St. Vincent’s Hospital Map Points of Interests 1. Registration -

Conferring of Awards 10, 11, 12 & 13 December 2019
CONFERRING OF AWARDS 10, 11, 12 & 13 DECEMBER 2019 Australian National Anthem Advance Australia Fair Australians all let us rejoice, For we are young and free; We’ve golden soil and wealth for toil; Our home is girt by sea; Our land abounds in nature’s gifts Of beauty rich and rare; In history’s page let every stage Advance Australia Fair. In joyful strains then let us sing, Advance Australia Fair. CONFERRING OF AWARDS Summer 2019 Llewellyn Hall The Australian National University Tuesday 10 December Wednesday 11 December Thursday 12 December Friday 13 December Chancellor: Professor the Honourable Gareth Evans AC QC BA LLB (Hons) Melb, MA Oxon, HonLLD Melb, Carleton, Syd FASSA Pro-Chancellor: Ms Naomi Flutter MPP Harvard, LLB (Hons), Bec (Hons), GDLP ANU Vice-Chancellor: Professor Brian P. Schmidt AC FAA FRS 2011 Nobel Laureate Physics BSc (Physics) Arizona, BSc (Astronomy) Arizona, MA (Astronomy) Harvard, PhD Harvard Esquire Bedel: Dr Ian Walker BA DipEd Syd, MA Macq, PhD UNSW University Marshal and Esquire Bedel: Ms Lorena Kanellopoulos DipHRM, GradCertMgt, MMgt ANU Mr Jake Francis Published by The Australian National University Conferring of Awards December 2019 1 CHANCELLOR’S MESSAGE TO GRADUANDS Today’s ceremony marks the culmination of years of research and study. ANU owes much to the intellectual and cultural contribution of our student body. In return, we work to build on our high standards in research and education. The ANU was created as part of a great nation building exercise in its day. That mandate continues and you share a vital part in it. -

Late Cretaceous) of Morocco : Palaeobiological and Behavioral Implications Remi Allemand
Endocranial microtomographic study of marine reptiles (Plesiosauria and Mosasauroidea) from the Turonian (Late Cretaceous) of Morocco : palaeobiological and behavioral implications Remi Allemand To cite this version: Remi Allemand. Endocranial microtomographic study of marine reptiles (Plesiosauria and Mosasauroidea) from the Turonian (Late Cretaceous) of Morocco : palaeobiological and behavioral implications. Paleontology. Museum national d’histoire naturelle - MNHN PARIS, 2017. English. NNT : 2017MNHN0015. tel-02375321 HAL Id: tel-02375321 https://tel.archives-ouvertes.fr/tel-02375321 Submitted on 22 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. MUSEUM NATIONAL D’HISTOIRE NATURELLE Ecole Doctorale Sciences de la Nature et de l’Homme – ED 227 Année 2017 N° attribué par la bibliothèque |_|_|_|_|_|_|_|_|_|_|_|_| THESE Pour obtenir le grade de DOCTEUR DU MUSEUM NATIONAL D’HISTOIRE NATURELLE Spécialité : Paléontologie Présentée et soutenue publiquement par Rémi ALLEMAND Le 21 novembre 2017 Etude microtomographique de l’endocrâne de reptiles marins (Plesiosauria et Mosasauroidea) du Turonien (Crétacé supérieur) du Maroc : implications paléobiologiques et comportementales Sous la direction de : Mme BARDET Nathalie, Directrice de Recherche CNRS et les co-directions de : Mme VINCENT Peggy, Chargée de Recherche CNRS et Mme HOUSSAYE Alexandra, Chargée de Recherche CNRS Composition du jury : M. -

P. 1 AC27 Inf. 7 (English Only / Únicamente En Inglés / Seulement
AC27 Inf. 7 (English only / únicamente en inglés / seulement en anglais) CONVENTION ON INTERNATIONAL TRADE IN ENDANGERED SPECIES OF WILD FAUNA AND FLORA ____________ Twenty-seventh meeting of the Animals Committee Veracruz (Mexico), 28 April – 3 May 2014 Species trade and conservation IUCN RED LIST ASSESSMENTS OF ASIAN SNAKE SPECIES [DECISION 16.104] 1. The attached information document has been submitted by IUCN (International Union for Conservation of * Nature) . It related to agenda item 19. * The geographical designations employed in this document do not imply the expression of any opinion whatsoever on the part of the CITES Secretariat or the United Nations Environment Programme concerning the legal status of any country, territory, or area, or concerning the delimitation of its frontiers or boundaries. The responsibility for the contents of the document rests exclusively with its author. AC27 Inf. 7 – p. 1 Global Species Programme Tel. +44 (0) 1223 277 966 219c Huntingdon Road Fax +44 (0) 1223 277 845 Cambridge CB3 ODL www.iucn.org United Kingdom IUCN Red List assessments of Asian snake species [Decision 16.104] 1. Introduction 2 2. Summary of published IUCN Red List assessments 3 a. Threats 3 b. Use and Trade 5 c. Overlap between international trade and intentional use being a threat 7 3. Further details on species for which international trade is a potential concern 8 a. Species accounts of threatened and Near Threatened species 8 i. Euprepiophis perlacea – Sichuan Rat Snake 9 ii. Orthriophis moellendorfi – Moellendorff's Trinket Snake 9 iii. Bungarus slowinskii – Red River Krait 10 iv. Laticauda semifasciata – Chinese Sea Snake 10 v. -
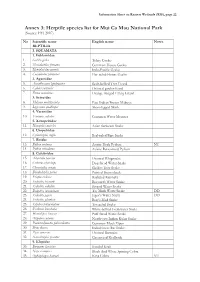
Information Sheet on Ramsar Wetlands (RIS), Page 22
Information Sheet on Ramsar Wetlands (RIS), page 22 Annex 3: Herptile species list for Mui Ca Mau National Park (Source: FFI 2007) No Scientific name English name Notes REPTILIA I. SQUAMATA 1. Gekkonidae 1. Gekko gecko Tokay Gecko 2. Hemidactylus frenatus Common House Gecko 3. Hemidactylus garnoti Indo-Pacific Gecko 4. Cosymbotus platyurus Flat-tailed House Gecko 2. Agamidae 5. Acanthosaura lepidogaster Scale-bellied Tree Lizard 6. Calotes versicolor Oriental garden lizard 7. Draco maculatus Orange-winged Flying Lizard 3. Scincidae 8. Mabuya multifasciata East Indian Brown Mabuya 9. Lygosoma quadrupes Short-legged Skink 4. Varanidae 10. Varanus salvator Common Water Monitor 5. Xenopeltidae 11. Xenopeltis unicolor Asian Sunbeam Snake 6. Uropeltidae 12. Cylindrophis ruffus Red-tailed Pipe Snake 7. Boidae 13. Python molurus Asiatic Rock Python NT 14. Python reticulatus Asiatic Reticulated Python 8. Colubridae 15. Ahaetulla prasina Oriental Whipsnake 16. Cerberus rhynchops Dog-faced Water Snake 17. Chrysopelea ornata Golden Tree Snake 18. Dendrelaphis pictus Painted Bronzeback 19. Elaphe radiata Radiated Ratsnake 20. Enhydris bocourti Bocourt's Water Snake 21. Enhydris enhydris Striped Water Snake 22. Enhydris innominata Tay Minh Water Snake DD 23. Enhydris jagori Jagor's Water Snake DD 24. Enhydris plumbea Boie's Mud Snake 25. Erpeton tentaculatum Tentacled Snake 26. Fordonia leucobalia White-bellied Freshwater Snake 27. Homalopsis buccata Puff-faced Water Snake 28. Oligodon cyclurus North-east Indian Kukri Snake 29. Psammodynastes pulverulentus Common Mock Viper 30. Ptyas korros Indochinese Rat Snake 31. Ptyas mucosus Oriental Ratsnake 32. Xenochrophis piscator Chequered Keelback 9. Elapidae 33. Bungarus fasciatus Banded Krait 34. Naja siamensis Black And White Spitting Cobra 35. -

Notice Warning Concerning Copyright Restrictions P.O
Publisher of Journal of Herpetology, Herpetological Review, Herpetological Circulars, Catalogue of American Amphibians and Reptiles, and three series of books, Facsimile Reprints in Herpetology, Contributions to Herpetology, and Herpetological Conservation Officers and Editors for 2015-2016 President AARON BAUER Department of Biology Villanova University Villanova, PA 19085, USA President-Elect RICK SHINE School of Biological Sciences University of Sydney Sydney, AUSTRALIA Secretary MARION PREEST Keck Science Department The Claremont Colleges Claremont, CA 91711, USA Treasurer ANN PATERSON Department of Natural Science Williams Baptist College Walnut Ridge, AR 72476, USA Publications Secretary BRECK BARTHOLOMEW Notice warning concerning copyright restrictions P.O. Box 58517 Salt Lake City, UT 84158, USA Immediate Past-President ROBERT ALDRIDGE Saint Louis University St Louis, MO 63013, USA Directors (Class and Category) ROBIN ANDREWS (2018 R) Virginia Polytechnic and State University, USA FRANK BURBRINK (2016 R) College of Staten Island, USA ALISON CREE (2016 Non-US) University of Otago, NEW ZEALAND TONY GAMBLE (2018 Mem. at-Large) University of Minnesota, USA LISA HAZARD (2016 R) Montclair State University, USA KIM LOVICH (2018 Cons) San Diego Zoo Global, USA EMILY TAYLOR (2018 R) California Polytechnic State University, USA GREGORY WATKINS-COLWELL (2016 R) Yale Peabody Mus. of Nat. Hist., USA Trustee GEORGE PISANI University of Kansas, USA Journal of Herpetology PAUL BARTELT, Co-Editor Waldorf College Forest City, IA 50436, USA TIFFANY -

Indicators Status and Trend Assessment
7/22/2015 BioRA Preparation Meeting Office of the Secretariat in Vientiane 04 – 11 July 2015 Frog and reptile Indicators Status and Trend Assessment Sereywath Pich Hoang Minh Duc Indicator: Ranid and Helid amphibian • Hylarana nigrovittata Status: • Unwell known, even in the IUCN list is just considered as least concerned species • Hylarana, for the last 15 Statusyears In Abundancethe Lower estimates Mekong as % relative River, to is2015 slightly Area declining 2015 1900 1950 1970 2000 Mekong• Hylarana River in Laosis under PDR threat Cby some123 factors% 123 % 123% 110% MekongThreats River in Laos PDR/Thailand C 123% 123% 123% 110% • Effort for food consumption Mekong• Loss River of habitat,in Cambodia forest canopyC over123 its% stream,123% caused123 %by destruction110% Tonleand Sap degradation River for agricultural,- -wood and- power plan- - Tonle Sap Great Lake - - - - - Mekong Delta - - - - - 1 7/22/2015 Indicator: Ranid and Helid amphibian Status:• Hoplobatrachus rugulosus • Tiger frog inhabits in various environment refugium, such as paddy field, irrigation infrastructure, fish pond, agricultural farm, ditch, floodplain wetland, forest pools, and other wet area Status Abundance estimates as % relative to 2015 • Tiger frog also encountered from sea level up to 700m asl. (Diesmos Area et al. 2004) 2015 1900 1950 1970 2000 • The global population, generally, is believed in stable condition Mekong River in Laos PDR C 123% 123% 123% 111% (Diesmos et al 2004) but it become rare in the lowland Mekong• Tiger River infrog Laos PDR/Thailand -

Boiga Cynodon • Characteristics
Photos of Common THAILAND SNAKES VERN LOVIC - THAILANDSNAKES.COM INTRODUCTION Welcome! Now that you’ve picked up this free ebook, share it with your friends. You can view it on almost any computer, smart phone, or tablet. It is available at the Apple iTunes store (free) and in PDF format at ThailandSnakes.com/ ebook/. This book covers what we believe to be the most common terrestrial (land-based) and freshwater snakes in Thailand, those you are likely to see - if you see any at all. We wrote this book to help educate the public and to hopefully save a few snakes from the shovel or machete. As you view the photos within, keep in mind that there are albino (no melanin) and melanistic (abundance of melanin) snakes that will not exhibit the same colors as most snakes of the species. Albino snakes can be pure white, or mostly white with a different colored pattern. Melanistic snakes are very dark, even solid black. So, you might see a white snake with a yellow pattern that looks exactly like the deadly Russell’s viper (Daboia russellii siamensis) - but the color is way off because it’s albino. It is still a deadly snake. We will release FREE UPDATES to this book in the future. If you haven’t signed up to be notified of updates - you won’t get them. Sign Up for Free Book Updates and Newsletter HERE (click red link) > If the link above does not work, visit: www.ThailandSnakes.com/ebook/ SNAKE BITE? Steps to take in the case of snake bite falls outside the scope of this ebook. -

Reproductive and Trophic Ecology of an Assemblage of Aquatic and Semi-Aquatic Snakes in Tonle Sap, Cambodia
Copeia 2009, No. 1, 7–20 Reproductive and Trophic Ecology of an Assemblage of Aquatic and Semi-Aquatic Snakes in Tonle Sap, Cambodia Sharon E. Brooks1, Edward H. Allison2, Jennifer A. Gill3, and John D. Reynolds4 We studied the reproductive and trophic ecology of a group of aquatic and semi-aquatic snakes that face severe hunting pressure in Cambodia. Over a two-year period we sampled hunters’ catches, measuring and dissecting a total of 8982 specimens of seven snake species, five of which belong to the family Homalopsidae. The seven species—Enhydris enhydris, Enhydris longicauda, Homalopsis buccata, Enhydris bocourti, Erpeton tentaculatus, Xenochrophis piscator, and Cylindrophis ruffus—all inhabit Tonle Sap Lake, the largest lake in South-East Asia. All species are sexually dimorphic in either body size or tail length. The larger species, E. bocourti and H. buccata, have a larger size at maturity, and the non- homalopsids, X. piscator and C. ruffus, have the highest and lowest fecundities, respectively. Clutch size increases significantly with female body size in all species, and with body conditioninE. enhydris. Our data also suggest that relative investment in reproduction increases with size in E. enhydris, which has the largest sample size. All species except one are synchronized in their timing of reproduction with the seasonally receding flood waters of the lake. There was variation in both the frequency of feeding and the prey size and type among species, with the homalopsids more similar to one another than to the other non-homalopsid species. The prey to predator mass ratio ranged from 0.04 to 0.1 in the homalopsids, compared to 0.15 to 0.17 in the non-homalopsids. -

RSB Director's Seminar Series
PUBLIC LECTURE RSB Director’s seminar series: Sex and speciation: did sex chromosome change trigger mammalian divergence? Friday 2 October 12.30–1.30pm with light lunch commencing 12pm Speaker Turnover of sex determining genes and Jenny Graves chromosomes may be a cataclysmic speciating event in many animals, in Professor, School of Life Science, contradiction of the accepted wisdom that La Trobe University speciation requires the accumulation of many small changes in isolated populations. There Location are many groups of invertebrates, fish and Slatyer Seminar Room reptiles in which closely related species have R.N. Robertson Building different sex determination mechanisms, and 46 Sullivans Creek Road interspecies hybridisation results in infertility. The Australian National University Here I propose that the evolution of the male- Contact determining SRY gene, defining a novel XY E [email protected] sex chromosome pair, was the event that T +61 2 6125 3841 interposed a reproductive barrier with the ancestral population of mammal-like reptiles, This lecture is free and open to the public and triggered the speciation event that was RSB event information: to lead to the evolution of therian mammals biology.anu.edu.au/news-events 190 million years ago. A sex chromosome-autosome fusion may, similarly, have separated eutherians (placental mammals) from marsupials 160 million years ago. A new burst of sex chromosome change and speciation seems to be be occurring in many mammal groups, precipitated by the degradation and disappearance of the mammalian Y chromosome. Although the primate Y seems to be unusually stable, sex chromosome turnover may ultimately promote hominid speciation. -

An Inconspicuous, Conspicuous New Species of Asian Pipesnake, Genus
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Wolverhampton Intellectual Repository and E-theses Zootaxa 4093 (1): 001–025 ISSN 1175-5326 (print edition) http://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2016 Magnolia Press ISSN 1175-5334 (online edition) http://doi.org/10.11646/zootaxa.4093.1.1 http://zoobank.org/urn:lsid:zoobank.org:pub:8C32F03F-E901-465D-B03D-7E6EEF288329 An inconspicuous, conspicuous new species of Asian pipesnake, genus Cylindrophis (Reptilia: Squamata: Cylindrophiidae), from the south coast of Jawa Tengah, Java, Indonesia, and an overview of the tangled taxonomic history of C. ruffus (Laurenti, 1768) MAX KIECKBUSCH1,4,§, SVEN MECKE1,§, LUKAS HARTMANN1, LISA EHRMANTRAUT1, MARK O’SHEA2 & HINRICH KAISER3 1Department of Animal Evolution and Systematics and Zoological Collection Marburg, Faculty of Biology, Philipps-Universität Mar- burg, Karl-von-Frisch-Straße 8, 35032 Marburg, Germany 2Faculty of Sciences and Engineering, University of Wolverhampton, Wulfruna Street, Wolverhampton, WV1 1LY, United Kingdom; and West Midland Safari Park, Bewdley, Worcestershire DY12 1LF, United Kingdom 3Department of Biology, Victor Valley College, 18422 Bear Valley Road, Victorville, California 92395, USA; and Department of Verte- brate Zoology, National Museum of Natural History, Smithsonian Institution, Washington, DC 20013, USA 4Corresponding author. E-mail: [email protected] §Co-first authors, listed in alphabetical order Abstract We describe a new species of Cylindrophis currently known only from Grabag, Purworejo Regency, Jawa Tengah Pro- vince (Central Java), Java, Indonesia. Cylindrophis subocularis sp. nov. can be distinguished from all congeners by the presence of a single, eponymous subocular scale between the 3rd and 4th or 4th and 5th supralabial, preventing contact be- tween the 4th or 5th supralabial and the orbit, and by having the prefrontal in narrow contact with or separated from the orbit. -

Bulletin and Proceedings 329 ABN 76 470 896 415 ISSN 1039-1843 September 2009
The Royal Society of New South Wales Bulletin and Proceedings 329 ABN 76 470 896 415 ISSN 1039-1843 September 2009 Future Events 2009 Lecture 7 October 2009, Darlington Centre at 7pm Lectures in Sydney are held in Lecture The SKAMP Project - a telescope reborn to look Room 1, Darlington Centre, University back in time of Sydney at 7 pm on the first Prof Anne Green, Head of School of Physics, University of Sydney Wednesday of the month with drinks available from 6 pm. or more than 40 years the University of Sydney has operated the Molonglo Wednesday 7 October 2009 7pm FObservatory. Recently, the Molonglo Observatory Synthesis Telescope completed The SKAMP Project - a a detailed imaging survey of the southern sky at a frequency of 843 MHz. What next? We are undertaking a complete renewal of the signal pathway as part of Australia’s telescope reborn to look contribution to the Square Kilometre Array (SKA) project, a powerful new radio back in time telescope. Our project is the SKA Molonglo Prototype (SKAMP), which will be a new Prof Anne Green low frequency spectrometer with wide-field imaging and polarisation capability. This Head of School of Physics talk will describe the project and how it builds on the previous telescope and its University of Sydney scientific achievements. Two of the key science goals to be undertaken initially will be a survey of red-shifted neutral hydrogen gas and a study of the transient radio sky. Friday 30 October 2009 5.30pm With the subsequent polarisation capability, we will map the magnetic field structure Clarke Memorial Lecture of our Galaxy and explore cosmic magnetism.