COFACTOR: an Accurate Comparative Algorithm for Structure-Based Protein Function Annotation Ambrish Roy, Jianyi Yang and Yang Zhang*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fruit Ripening and Storage
OPEN Citation: Horticulture Research (2014) 1, 6; doi:10.1038/hortres.2014.6 ß 2014 Nanjing Agricultural University All rights reserved 2052-7276/14 www.nature.com/hortres ARTICLE Dynamic changes in proteins during apple (Malus x domestica) fruit ripening and storage Yun Shi1, Li Jiang1, Li Zhang2, Ruoyi Kang1 and Zhifang Yu1 A proteomic study, using two-dimensional polyacrylamide gel electrophoresis and matrix-assisted laser desorption/ionization time-of-flight/time-of-flight, was conducted in apple fruit (cv. ‘Golden Delicious’) starting at 10 days prior to harvest through 50 days in storage. Total protein was extracted using a phenol/sodium dodecyl sulfate protocol. More than 400 protein spots were detected in each gel and 55 differentially expressed proteins (p,0.05) were subjected to matrix-assisted laser desorption/ionization time-of-flight/ time-of-flight analysis. Fifty-three of these proteins were finally identified using an apple expressed sequence tag database downloaded from Genome Database for Rosaceae and placed into six categories. The categories and the percentage of proteins placed in each category were stress response and defense (49.0%), energy and metabolism (34.0%), fruit ripening and senescence (5.6%), signal transduction (3.8%), cell structure (3.8%) and protein synthesis (3.8%). Proteins involved in several multiple metabolic pathways, including glycolysis, pentose–phosphate pathway, anti-oxidative systems, photosynthesis and cell wall synthesis, were downregulated, especially during the climacteric burst in respiration and during the senescent stages of fruit development. Proteins classified as allergens or involved in cell wall degradation were upregulated during the ripening process. Some protein spots exhibited a mixed pattern (increasing to maximal abundance followed by a decrease), such as 1-aminocyclopropane-1-carboxylate oxidase, L-ascorbate peroxidase and abscisic acid response proteins. -

Genome-Wide Analysis of Glyoxalase-Like Gene Families in Grape
Li et al. BMC Genomics (2019) 20:362 https://doi.org/10.1186/s12864-019-5733-y RESEARCHARTICLE Open Access Genome-wide analysis of glyoxalase-like gene families in grape (Vitis vinifera L.) and their expression profiling in response to downy mildew infection Tiemei Li1,2,3, Xin Cheng1,2,3, Yuting Wang1,2,3, Xiao Yin1,2,3, Zhiqian Li1,2,3, Ruiqi Liu1,2,3, Guotian Liu1,2,3, Yuejin Wang1,2,3 and Yan Xu1,2,3* Abstract Background: The glyoxalase system usually comprises two enzymes, glyoxalase I (GLYI) and glyoxalase II (GLYII). This system converts cytotoxic methylglyoxal (MG) into non-toxic D-lactate in the presence of reduced glutathione (GSH) in two enzymatic steps. Recently, a novel type of glyoxalase III (GLYIII) activity has observed in Escherichia coli that can detoxify MG into D-lactate directly, in one step, without a cofactor. Investigation of the glyoxalase enzymes of a number of plant species shows the importance of their roles in response both to abiotic and to biotic stresses. Until now, glyoxalase gene families have been identified in the genomes of four plants, Arabidopsis, Oryza sativa, Glycine max and Medicago truncatula but no similar study has been done with the grapevine Vitis vinifera L. Results: In this study, four GLYI-like,twoGLYII-like and three GLYIII-like genesareidentifiedfromthegenomedatabaseof grape. All these genes were analysed in detail, including their chromosomal locations, phylogenetic relationships, exon-intron distributions, protein domain organisations and the presence of conserved binding sites. Using quantitative real-time PCR analysis (qRT-PCR), the expression profiles of these geneswereanalysedindifferent tissues of grape, and also when under infection stress from downy mildew (Plasmopara viticola). -
Supplementary File 1
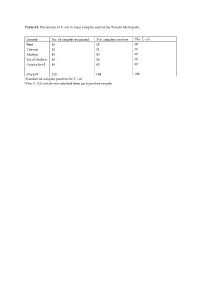
Table S1. Prevalence of E. coli in meat samples sold at the Tamale Metropolis. Sample No. of samples examined aNo. samples positive bNo. E. coli Beef 45 39 39 Chevon 45 34 34 Mutton 45 40 40 Local chicken 45 36 36 Guinea fowl 45 40 40 Overall 225 189 189 aNumber of samples positive for E. coli. bOne E. Coli isolate was selected from each positive sample. Table S2. A table showing the eBURST (Based Upon Related Sequence Types) analyses of the study sequence types with global curated STs in Escherichia PubMLST database. MLST (Isolate) Type of clone Closet global ancestry Source sequence type (ST) ST69 (SG6) Similar a ST69 Animal (Food), Human ST155 (SLC2, Similar ST155 Animal (Food), Human, TLC13, CM4) Environment ST297 (TLC1) Similar ST297 Human ST1727 (NC3) Similar ST1727 Human ST44 (AC1) Single-Locus Variant ST10, ST752 Animal (Food), (SLV) b Human ST469 (CC6) Single-Locus Variant ST162 Food (SLV) ST540 (AB1, Single-Locus Variant ST4093 Human TG1) (SLV) ST1141 (NM11) Single-Locus Variant ST10, ST744 Animal (Food), (SLV) Human ST7473 (NB12) Single-Locus Variant ST10 Animal (Food), (SLV) Human ST6646 (CB1) Satellite c None - ST7483 (NB12) Satellite None - a Similar: study isolate was similar to a global curated known sequence type. b Single-Locus Variant (SLV): study isolate only shared similarity with global curated known sequence types that differed in one allelic gene. c Satellite: study isolate as a distantly related and did not shared any similarity with global curated known sequence types. Table S3. In silico identification and characterization of conserved stress response mechanisms in the E. -

Supplementary Information 2 to Accompany
1 Supplementary Information 2 to accompany 3 Sulfur-oxidizing symbionts without canonical genes for autotrophic CO2 fixation 4 Brandon K. B. Seah*1,7, Chakkiath Paul Antony1,8, Bruno Huettel2, Jan Zarzycki3, Lennart 5 Schada von Borzyskowski3, Tobias J. Erb3, Angela Kouris4, Manuel Kleiner5, Manuel 6 Liebeke1, Nicole Dubilier1,6, Harald R. Gruber-Vodicka1 7 1 Max Planck Institute for Marine Microbiology, Celsiusstraße 1, 28359 Bremen, Germany 8 2 Max Planck Genome Centre Cologne, Max Planck Institute for Plant Breeding Research, 9 Carl-von-Linné-Weg 10, 50829 Cologne, Germany 10 3 Max Planck Institute for Terrestrial Microbiology, Karl-von-Frisch-Str. 10, 35043 Marburg, 11 Germany 12 4 Energy Bioengineering and Geomicrobiology Group, University of Calgary, 2500 13 University Drive Northwest, Calgary, Alberta T2N 1N4, Canada 14 5 Department of Plant and Microbial Biology, North Carolina State University, Raleigh 15 27695, North Carolina, United States of America 16 6 MARUM, Center for Marine Environmental Sciences, University of Bremen, 28359 17 Bremen, Germany 18 7 Current address: Max Planck Institute for Developmental Biology, Max-Planck-Ring 5, 19 72076 Tübingen, Germany 20 8 Current address: Red Sea Research Center, Biological and Environmental Sciences and 21 Engineering (BESE) Division, King Abdullah University of Science and Technology 22 (KAUST), Thuwal 23955, Kingdom of Saudi Arabia 23 * Corresponding author 1 24 Supplementary Materials and Methods 25 Metabolite extraction and identification 26 Kentrophoros sp. H was collected on Elba in 2014 for metabolomics (Supplementary Table 27 7). Samples were fixed in 1 mL cold methanol (HPLC-grade, Sigma-Aldrich) and stored at - 28 20°C until use. -

Recombinant Human Glyoxalase I Catalog Number: 4959-GL
Recombinant Human Glyoxalase I Catalog Number: 4959-GL DESCRIPTION Source E. coliderived Ala2Met184, with an Nterminal Met and 6His tag Accession # NP_006699 Nterminal Sequence Met Analysis Predicted Molecular 22 kDa Mass SPECIFICATIONS SDSPAGE 25 kDa, reducing conditions Activity Measured by its ability to catalyze the formation of SDlactoylglutathione from the hemimercaptal adduct that forms spontaneously between methylglyoxal and reduced glutathione. The specific activity is >100 nmol/min/µg, as measured under the described conditions. Endotoxin Level <1.0 EU per 1 μg of the protein by the LAL method. Purity >85%, by SDSPAGE under reducing conditions and visualized by silver stain. Formulation Lyophilized from a 0.2 μm filtered solution in TrisHCl and DTT. See Certificate of Analysis for details. Activity Assay Protocol Materials l Assay Buffer: 0.1 M Sodium Phosphate, pH 7.0 l Recombinant Human Glyoxalase I (rhGlyoxalase I) (Catalog # 4959GL) l Glutathione, Reduced (GSH) (Amresco, Catalog # 0399) l Methylglyoxal solution, 40% (Sigma, Catalog # M0252) l 96well Clear UV Plate (Costar, Catalog # 3635) l Plate Reader (Model: SpectraMax Plus by Molecular Devices) or equivalent Assay 1. Prepare 100 mM GSH in deionized water. Note: Prepare fresh. 2. Dilute 40% (6.48 M) Methylglyoxal solution to 100 mM in Assay Buffer. Note: Prepare fresh. 3. Combine 1420 µL Assay Buffer, 40 µL 100 mM GSH, and 40 µL 100 mM Methylglyoxal to make the Substrate Mixture. 4. Incubate at room temperature for 15 minutes. 5. Dilute rhGlyoxalase I to 0.4 ng/µL in Assay Buffer. -

Glyoxalase Activity in Human Erythrocytes and Mouse Lymphoma, Liver and Brain Probed with Hyperpolarized C-13-Methylglyoxal Dmitry Shishmarev, Philip W
Glyoxalase activity in human erythrocytes and mouse lymphoma, liver and brain probed with hyperpolarized C-13-methylglyoxal Dmitry Shishmarev, Philip W. Kuchel, Guilhem Pages, Alan J. Wright, Richard L. Hesketh, Felix Kreis, Kevin M. Brindle To cite this version: Dmitry Shishmarev, Philip W. Kuchel, Guilhem Pages, Alan J. Wright, Richard L. Hesketh, et al.. Glyoxalase activity in human erythrocytes and mouse lymphoma, liver and brain probed with hyperpolarized C-13-methylglyoxal. Communications Biology, Nature Publishing Group, 2018, 1, 10.1038/s42003-018-0241-1. hal-02624539 HAL Id: hal-02624539 https://hal.inrae.fr/hal-02624539 Submitted on 26 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ARTICLE https://doi.org/10.1038/s42003-018-0241-1 OPEN Glyoxalase activity in human erythrocytes and mouse lymphoma, liver and brain probed with hyperpolarized 13C-methylglyoxal Dmitry Shishmarev 1, Philip W. Kuchel2, Guilhem Pagès 3, Alan J. Wright 4, Richard L. Hesketh4, 1234567890():,; Felix Kreis 4 & Kevin M. Brindle 4 Methylglyoxal is a faulty metabolite. It is a ubiquitous by-product of glucose and amino acid metabolism that spontaneously reacts with proximal amino groups in proteins and nucleic acids, leading to impairment of their function. -

Mobilization of Iron Stored in Bacterioferritin Is Required for Metabolic Homeostasis in Pseudomonas Aeruginosa
SUPPORTING INFORMATION Mobilization of Iron Stored in Bacterioferritin is Required for Metabolic Homeostasis in Pseudomonas aeruginosa Achala N. D. Punchi Hewage 1, Leo Fontenot 2, Jessie Guidry 3, Thomas Weldeghiorghis 2, Anil K. Mehta 4, Fabrizio Donnarumma 2, and Mario Rivera 2,* 1 Department of Chemistry, University of Kansas, 2030 Becker Dr., Lawrence, KS, 66047, USA; [email protected] 2 Department of Chemistry, Louisiana State University, 232 Choppin Hall, Baton Rouge, LA, 70803, USA; [email protected] (L.F); [email protected] (T.W); [email protected] (F.D) 3 Department of Biochemistry and Molecular Biology, Louisiana State University Health Science Center, 1901 Perdido Street, New Orleans, LA, 70112, USA; [email protected] 4 National High Magnetic Field Laboratory, University of Florida, 1149 Newell Drive, Gainesville, FL, 32610, USA; [email protected]. *Corresponding author. E-mail: [email protected] ORCID: 0000-0002-5692-5497 Figure S1. Growth curves and levels of pyoverdine secreted by wt and Δbfd P. aeruginosa cells. (A) P. aeruginosa cells (wt and Δbfd) were cultured in PI media supplemented with 10 µM Fe at 37 °C and shaking at 220 rpm. For the purpose of all the analyses reported in this work, the cells were harvested by centrifugation 30 h post inoculation. (B) Pyoverdine secreted by the cells was measured in the cell-free supernatants by acquiring fluorescence emission spectra (430-550 nm) with excitation at 400 nm (10 nm slit width) and emission at 460 nm (10 nm slit width). Fluorescence intensity normalized to viable cell count (CFU/mL) shows that the Δbfd cells secrete approximately sixfold more pyoverdine than the wt cells. -

Electronic Supplementary Information S9
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2019 Electronic Supplementary Information S9 . Changes in gene expression due to As III exposure in the contrasting hairy roots. HYBRIDIZATION 1: LIST OF UP-REGULATED GENES Fold Change Probe Set ID ([0.1.HR] vs [0.HR]) Blast2GO description Genbank Accessions Serine/threonine -protein phosphatase 7 long form BT2M444_at 101.525 homolog AJ538927 DV159578_at 67.016 Retrotransposon Tnt1 s231d long terminal repeat DV159578 EB427540_at 49.6314 DNA repair metallo-beta-lactamase family protein EB427540 C3356_at 39.8605 Late embryogenesis abundant domain-containing protein DV157779 C7375_at 39.0452 wall-associated serine threonine kinase BP525596 C11724_at 37.103 5' exonuclease Apollo-like, transcript variant 2 AJ718369 EB682553_at 36.7071 EIF4A-2 EB682553 U92011_at 34.5134 apocytochrome b U92011 C5842_at 33.8783 heat shock protein 90 EB432766 AF211619_at 31.8765 AF211619 BC1M4448_s_at 31.2217 AJ538430 TT39_L17_at 29.7963 lysine-ketoglutarate reductase EB429701_at 29.3945 EB429701 BP137485_at 29.0749 Retrotransposon Tnt1 s231d long terminal repeat BP137485 CV021716_at 28.8452 Plastocyanin-like domain-containing protein CV021716 DW002835_at 27.8539 peroxidase DW002835 EB429725_at 27.6171 Carbonic anhydrase, transcript variant 1 (ca1), mRNA EB429725 TT19_B11_at 26.3284 50s ribosomal protein l16 DV158314_at 24.4108 DV158314 C7180_at 23.668 cytochrome oxidase subunit i BP528067 EB445091_at 22.6679 lysine-ketoglutarate reductase EB445091 GC227C_at -

Modeling and Computational Prediction of Metabolic Channelling
MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING by Christopher Morran Sanford A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Molecular Genetics University of Toronto © Copyright by Christopher Morran Sanford 2009 Abstract MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING Master of Science 2009 Christopher Morran Sanford Graduate Department of Molecular Genetics University of Toronto Metabolic channelling occurs when two enzymes that act on a common substrate pass that intermediate directly from one active site to the next without allowing it to diffuse into the surrounding aqueous medium. In this study, properties of channelling are investigated through the use of computational models and cell simulation tools. The effects of enzyme kinetics and thermodynamics on channelling are explored with the emphasis on validating the hypothesized roles of metabolic channelling in living cells. These simulations identify situations in which channelling can induce acceleration of reaction velocities and reduction in the free concentration of intermediate metabolites. Databases of biological information, including metabolic, thermodynamic, toxicity, inhibitory, gene fusion and physical protein interaction data are used to predict examples of potentially channelled enzyme pairs. The predictions are used both to support the hypothesized evolutionary motivations for channelling, and to propose potential enzyme interactions that may be worthy of future investigation. ii Acknowledgements I wish to thank my supervisor Dr. John Parkinson for the guidance he has provided during my time spent in his lab, as well as for his extensive help in the writing of this thesis. I am grateful for the advice of my committee members, Prof. -

Over-Expression and Characterization of a Glyoxalase 2 Like Enzyme
MIAMI UNIVERSITY The Graduate School Certificate for Approving the Dissertation We hereby approve the Dissertation of Pattraranee Limphong Candidate for the Degree: Doctor of Philosophy _______________________________________________ Director Dr. Christopher A. Makaroff _______________________________________________ Reader Dr. Michael W. Crowder _______________________________________________ Reader Dr. John W. Hawes _______________________________________________ Reader Dr. Neil D. Danielson _______________________________________________ Graduate School Representative Dr. Qingshun Li ABSTRACT OVER-EXPRESSION AND CHARACTERIZATION OF A GLYOXALASE 2 LIKE ENZYME By Pattraranee Limphong This dissertation consists of seven chapters to better probe and understand the structure and function of a glyoxalase 2 (GLX2) like enzyme from Arabidopsis thaliana and human. Chapter 1 provides a general background of the glyoxalase system followed by the physiological roles of the glyoxalase system, and inhibitors of the glyoxalase enzymes, glyoxalase 1 and glyoxalase 2. Chapter 2 focuses on over-expression and characterization of GLX2-1 (putative glyoxalase 2, isozyme1) from Arabidopsis thaliana. Metal analyses, kinetics, and spectroscopic studies suggested that GLX2-1 contains a dinuclear metal binding site but is not a glyoxalase 2. Chapter 3 investigates the physiological role of GLX2-1. A commercial survey substrate system and β-lactamase substrates were used. The results showed that GLX2-1 exhibits β-lactamase activity. Chapter 4 presents the results of experiments to convert inactive GLX2-1 into an active GLX2 enzyme. Substrate binding residues were mutated at positions 225, 253, 255, 332, and 335 in GLX2-1. The results showed that the R253H GLX2-1 does hydrolyze S-D-lactoylglutathione (SLG) when the substrate binding ligands were generated on the R253H GLX2-1 enzyme. Chapter 5 provides detailed structural information on the human GLX2 metal center and insights concerning the structure and kinetic mechanism of the enzyme. -

Genome-Scale Metabolic Network Analysis and Drug Targeting of Multi-Drug Resistant Pathogen Acinetobacter Baumannii AYE
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2017 Electronic Supplementary Information (ESI) for Molecular BioSystems Genome-scale metabolic network analysis and drug targeting of multi-drug resistant pathogen Acinetobacter baumannii AYE Hyun Uk Kim, Tae Yong Kim and Sang Yup Lee* E-mail: [email protected] Supplementary Table 1. Metabolic reactions of AbyMBEL891 with information on their genes and enzymes. Supplementary Table 2. Metabolites participating in reactions of AbyMBEL891. Supplementary Table 3. Biomass composition of Acinetobacter baumannii. Supplementary Table 4. List of 246 essential reactions predicted under minimal medium with succinate as a sole carbon source. Supplementary Table 5. List of 681 reactions considered for comparison of their essentiality in AbyMBEL891 with those from Acinetobacter baylyi ADP1. Supplementary Table 6. List of 162 essential reactions predicted under arbitrary complex medium. Supplementary Table 7. List of 211 essential metabolites predicted under arbitrary complex medium. AbyMBEL891.sbml Genome-scale metabolic model of Acinetobacter baumannii AYE, AbyMBEL891, is available as a separate file in the format of Systems Biology Markup Language (SBML) version 2. Supplementary Table 1. Metabolic reactions of AbyMBEL891 with information on their genes and enzymes. Highlighed (yellow) reactions indicate that they are not assigned with genes. No. Metabolism EC Number ORF Reaction Enzyme R001 Glycolysis/ Gluconeogenesis 5.1.3.3 ABAYE2829 -

Over-Expression and Characterization of a Metallohydrolase from Arabidopsis Thaliana
ABSTRACT GLYOXALASE 2-2: OVER-EXPRESSION AND CHARACTERIZATION OF A METALLOHYDROLASE FROM ARABIDOPSIS THALIANA By Nathan F. Wenzel In order to investigate the metal binding properties of A. thaliana glyoxalase 2-2, recombinant GLX 2-2 was over-expressed in rich or minimal media containing various amounts of added Zn(II), Fe(II), or Mn(II). The resulting enzymes were purified, characterized, and compared. Kinetic studies and metal analyses of the samples indicated that no significant catalytic differences existed between the enzymes, in spite of the enzymes binding different types and amounts of metal ions. EPR and EXAFS spectroscopies strongly indicated the presence of positive cooperativity of metal binding and of numerous possible dinuclear metal centers, including Fe(III)Zn(II), Fe(III)Fe(II), and Mn(II)Mn(II) centers. The ability to bind various amounts of manganese, iron, and zinc with no apparent change in catalytic activity and the possibility of positive cooperativity in metal binding make A. thaliana glyoxalase 2-2 unique to the family of proteins containing the β-lactamase fold. Glyoxalase 2-2: Over-expression and Characterization of a Metallohydrolase from Arabidopsis thaliana A Thesis Submitted to the Faculty of Miami University In partial fulfillment of The requirement for the degree of Master of Science Department of Chemistry and Biochemistry By Nathan F. Wenzel Miami University Oxford, OH 2003 Advisor________________________ Michael W. Crowder Reader_________________________ Christopher A. Makaroff Reader_________________________ John W. Hawes Reader_________________________ Robert E. Minto Table of Contents 1. Chapter 1 1.1. Introduction: the Glyoxalase system 1 1.2. Physiological Roles of the Glyoxalase system 3 1.3.