Origin and Evolution of Deleterious Mutations in Horses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pferdezucht, -Haltung Und -Fütterung Empfehlungen Für Die Praxis
Sonderheft 353 Special Issue Pferdezucht, -haltung und -fütterung Empfehlungen für die Praxis Wilfried Brade, Ottmar Distl, Harald Sieme und Anette Zeyner (Hrsg.) Landbauforschung vTI Agriculture and Forestry Research Sonderheft 353 Special Issue Preis / Price 10 € LBF_SH_353_U4 9,63 LBF_SH_353_U1 Bibliografische Information der Deutschen Bibliothek Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen Nationalbiblio- grafie; detaillierte bibliografische Daten sind im Internet über http:// www.d-nb.de/ abrufbar. 2011 Landbauforschung vTI Agriculture and Forestry Research Johann Heinrich von Thünen-Institut Bundesforschungsinstitut für Ländliche Räume, Wald und Fischerei (vTI) Bundesallee 50, D-38116 Braunschweig, Germany Die Verantwortung für die Inhalte liegt bei den jeweiligen Verfassern bzw. Verfasserinnen. [email protected] www.vti.bund.de Preis 10 € ISSN 0376-0723 ISBN 978-3-86576-079-1 LBF_SH 353_U2 9,63 LBF_SH 353_U3 Landbauforschung vTI Agriculture and Forestry Research Sonderheft 353 Special Issue Pferdezucht, -haltung und -fütterung Empfehlungen für die Praxis Wilfried Brade1, Ottmar Distl2, Harald Sieme3 und Anette Zeyner4 (Hrsg.) 1 Stiftung Tierärztl. Hochschule Hannover, zur Zeit: Leibnitz-Institut für Nutztierbiologie (FBN), Wilhelm-Stahl-Allee 2, 18196 Dummerstorf, Email: [email protected] 2 Stiftung Tierärztl. Hochschule Hannover, Institut für Tierzucht und Vererbungsfor- schung, Bünteweg 17 p, 30559 Hannover, Email: [email protected] 3 Stiftung Tierärztl. Hochschule Hannover, Reprod.-med. Einheit der Kliniken, Bünteweg 15, 30559 Hannover, Email: [email protected] 4 Universität Rostock, Professur für Ernährungsphysiologie u. Tierernährung, Justus- von-Liebig-Weg 8, 18059 Rostock, Email: [email protected] W. Brade, O. Distl, H. Sieme, A. Zeyner (Hrsg.), Pferdezucht, -haltung und -fütterung - Empfehlungen für die Praxis Vorwort Lange Zeit befanden sich die meisten Pferde in bäuerlicher Hand. -

CHA TM CHA–THE LEADERS in HORSEMANSHIP SAFETY Changing Lives Through Safe Experiences with Horses •
CHA Certified Horsemanship Association International Conference September 28 – 30, 2018 Colorado State University Equine Sciences CHA TM CHA–THE LEADERS IN HORSEMANSHIP SAFETY Changing Lives Through Safe Experiences with Horses WWW.CHA.HORSE • WWW.CHAINSTRUCTORS.COM CHA CORPORATE OFFICE & STAFF NEW AD RATES 1795 Alysheba Way Ste. 7102 | Lexington, KY 40509 THE INSTRUCTOR 859-259-3399 | [email protected] www.CHA.horse | www.CHAinstructors.com MAGAZINE Christy Landwehr–Chief Executive Officer | 720-857-9550 | [email protected] We print 4,500 issues Terri Weaver–Membership Services Director | 859-259-3399 | [email protected] that are distributed to all Julie Goodnight–International Spokesperson | 719-530-0531 | [email protected] CHA members, CHA individual and equine Executive Committee Board of Directors facilities program members and at our In- Elizabeth Duffy .............................. [email protected] President–Beth Powers ........................ [email protected] Jennifer Eaton ............................... [email protected] ternational and Regional President Elect–Tammi Gainer [email protected] Hayley Eberle ............................. [email protected] Conferences and at all Vice Pres. of Reg. Relations–Anne Brzezicki [email protected] Tara Gamble [email protected] trade shows and events Vice Pres. of New Initiatives–Robert Coleman [email protected] Christine Gillett [email protected] Shellie Hensley -

FEI Regulations for Equestrian Events at the Olympic Games
FEI Fédération Equestre Internationale FEI Regulations for Equestrian Events at the Olympic Games 24th Edition, Effective for the Olympic Games Tokyo 2020 23 July-8 August 2021 Fédération Equestre Internationale t +41 21 310 47 47 HM King Hussein I Building f +41 21 310 47 60 Chemin de la Joliette 8 www.fei.org 1006 Lausanne Switzerland Printed in Switzerland Copyright © 2018 Fédération Equestre Internationale 7 December 2018 Updated on 21 December 2018 Updated on 30 December 2018 Updated on 18 April 2019 Updated on 3 October 2019 Updated on 24 June 2020 Updated on 16 June 2021 FEI Regulations for Equestrian Events Tokyo (JPN) 2020 Olympic Games TABLE OF CONTENTS THE FEI CODE OF CONDUCT FOR THE WELFARE OF THE HORSE .................................. 4 CHAPTER I GENERAL .................................................................................................. 6 ARTICLE 600 – INTRODUCTION .................................................................................................. 6 ARTICLE 601 –COMPETITIONS .................................................................................................... 6 ARTICLE 602 – COMPETITION SCHEDULE .................................................................................... 7 ARTICLE 603 – CLASSIFICATION, MEDALS & PRIZES..................................................................... 7 ARTICLE 604 – QUOTA .............................................................................................................. 8 ARTICLE 605 - AP ALTERNATE ATHLETES, RESERVE HORSES, -

Essential Genetics for the Horseman
Essential Genetics for the Horseman Whether you are an owner, breeder, rider or other equine enthusiast, there are some essential genetics that can help you to make the most of your journey. This primer is meant to introduce you to the basics of inheritance. Over the last decade, scientific research in equine genetics and the mapping of the equine genome has led to a number of interesting and valuable discoveries. Some of these are directly relevant to the Arabian breed, especially the discoveries relating to disease genes. However, a number of very interesting insights about the entire equine species have been discovered. DNA and Genes The basic starting material for genetics in all species is their DNA. The horse genome (the collection of all the DNA in each cell of the horse) has been completely sequenced, just like in the human. The genome of the horse consists of about 2.7 billion base pairs (the basic unit of DNA, abbreviated by G, A, T, C). This is in comparison to the human genome that has just over 3.0 billion base pairs. When these base pairs are strung together in sets of a few thousand at a time, they provide the ‘blueprint’ for about 20,000 different genes in the horse. About 17,000 of these genes are very similar both in sequence and in function to the corresponding human gene. Chromosomes These genes are physically located on 64 chromosomes in 32 pairs, located in every cell of the horse. Each foal receives a set of 32 chromosomes containing 20,000 genes from its’ dam and also a set from its’ sire. -

Identification of Copy Number Variants in Horses
Downloaded from genome.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Research Identification of copy number variants in horses Ryan Doan,1 Noah Cohen,2 Jessica Harrington,2 Kylee Veazy,2 Rytis Juras,3 Gus Cothran,3 Molly E. McCue,4 Loren Skow,3 and Scott V. Dindot1,5,6 1Department of Veterinary Pathobiology, 2Department of Large Animal Clinical Sciences, 3Department of Veterinary Integrative Biosciences, College of Veterinary Medicine and Biomedical Sciences, Texas A&M University, College Station, Texas 77843, USA; 4Department of Veterinary Population Medicine, University of Minnesota College of Veterinary Medicine, St. Paul, Minnesota 55108, USA; 5Department of Molecular and Cellular Medicine, Texas A&M Health Science Center College of Medicine, College Station, Texas 77843, USA Copy number variants (CNVs) represent a substantial source of genetic variation in mammals. However, the occurrence of CNVs in horses and their subsequent impact on phenotypic variation is unknown. We performed a study to identify CNVs in 16 horses representing 15 distinct breeds (Equus caballus) and an individual gray donkey (Equus asinus) using a whole- exome tiling array and the array comparative genomic hybridization methodology. We identified 2368 CNVs ranging in size from 197 bp to 3.5 Mb. Merging identical CNVs from each animal yielded 775 CNV regions (CNVRs), involving 1707 protein- and RNA-coding genes. The number of CNVs per animal ranged from 55 to 347, with median and mean sizes of CNVs of 5.3 kb and 99.4 kb, respectively. Approximately 6% of the genes investigated were affected by a CNV. Biological process enrichment analysis indicated CNVs primarily affected genes involved in sensory perception, signal transduction, and metabolism. -

List of Horse Breeds 1 List of Horse Breeds
List of horse breeds 1 List of horse breeds This page is a list of horse and pony breeds, and also includes terms used to describe types of horse that are not breeds but are commonly mistaken for breeds. While there is no scientifically accepted definition of the term "breed,"[1] a breed is defined generally as having distinct true-breeding characteristics over a number of generations; its members may be called "purebred". In most cases, bloodlines of horse breeds are recorded with a breed registry. However, in horses, the concept is somewhat flexible, as open stud books are created for developing horse breeds that are not yet fully true-breeding. Registries also are considered the authority as to whether a given breed is listed as Light or saddle horse breeds a "horse" or a "pony". There are also a number of "color breed", sport horse, and gaited horse registries for horses with various phenotypes or other traits, which admit any animal fitting a given set of physical characteristics, even if there is little or no evidence of the trait being a true-breeding characteristic. Other recording entities or specialty organizations may recognize horses from multiple breeds, thus, for the purposes of this article, such animals are classified as a "type" rather than a "breed". The breeds and types listed here are those that already have a Wikipedia article. For a more extensive list, see the List of all horse breeds in DAD-IS. Heavy or draft horse breeds For additional information, see horse breed, horse breeding and the individual articles listed below. -

Biodiversity of Arabian Horses in Syria
Biodiversity of Arabian horses in Syria Dissertation zur Erlangung des akademischen Grades Doctor rerum agriculturarum (Dr. rer. agr.) eingereicht an der Lebenswissenschaftlichen Fakultät der Humboldt Universität zu Berlin von M.Sc. Saria Almarzook Präsidentin der Humboldt-Universität zu Berlin Prof. Dr. Sabine Kunst Dekan der Humboldt-Universität zu Berlin Prof. Dr. Bernhard Grimm Gutachterin/Gutachter Prof. Dr. Gudrun Brockmann Prof. Dr. Dirk Hinrichs Prof. Dr. Armin Schmitt Tag der mündlichen Prüfung: 17. September 2018 Dedication This research is dedicated to my homeland …Syria Contents Zusammenfassung ................................................................................................................... I Summary ............................................................................................................................... VI List of publications and presentations .................................................................................. XII List of abbreviations ............................................................................................................ XIII List of figures ....................................................................................................................... XIV List of tables ......................................................................................................................... XV 1. General introduction and literature review ..................................................................... 1 1.1. Domestication and classification -
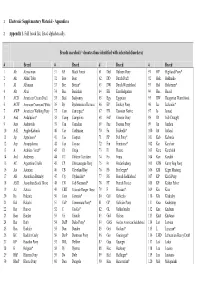
Electronic Supplementary Material - Appendices
1 Electronic Supplementary Material - Appendices 2 Appendix 1. Full breed list, listed alphabetically. Breeds searched (* denotes those identified with inherited disorders) # Breed # Breed # Breed # Breed 1 Ab Abyssinian 31 BF Black Forest 61 Dul Dülmen Pony 91 HP Highland Pony* 2 Ak Akhal Teke 32 Boe Boer 62 DD Dutch Draft 92 Hok Hokkaido 3 Al Albanian 33 Bre Breton* 63 DW Dutch Warmblood 93 Hol Holsteiner* 4 Alt Altai 34 Buc Buckskin 64 EB East Bulgarian 94 Huc Hucul 5 ACD American Cream Draft 35 Bud Budyonny 65 Egy Egyptian 95 HW Hungarian Warmblood 6 ACW American Creme and White 36 By Byelorussian Harness 66 EP Eriskay Pony 96 Ice Icelandic* 7 AWP American Walking Pony 37 Cam Camargue* 67 EN Estonian Native 97 Io Iomud 8 And Andalusian* 38 Camp Campolina 68 ExP Exmoor Pony 98 ID Irish Draught 9 Anv Andravida 39 Can Canadian 69 Fae Faeroes Pony 99 Jin Jinzhou 10 A-K Anglo-Kabarda 40 Car Carthusian 70 Fa Falabella* 100 Jut Jutland 11 Ap Appaloosa* 41 Cas Caspian 71 FP Fell Pony* 101 Kab Kabarda 12 Arp Araappaloosa 42 Cay Cayuse 72 Fin Finnhorse* 102 Kar Karabair 13 A Arabian / Arab* 43 Ch Cheju 73 Fl Fleuve 103 Kara Karabakh 14 Ard Ardennes 44 CC Chilean Corralero 74 Fo Fouta 104 Kaz Kazakh 15 AC Argentine Criollo 45 CP Chincoteague Pony 75 Fr Frederiksborg 105 KPB Kerry Bog Pony 16 Ast Asturian 46 CB Cleveland Bay 76 Fb Freiberger* 106 KM Kiger Mustang 17 AB Australian Brumby 47 Cly Clydesdale* 77 FS French Saddlebred 107 KP Kirdi Pony 18 ASH Australian Stock Horse 48 CN Cob Normand* 78 FT French Trotter 108 KF Kisber Felver 19 Az Azteca -

Eric Lamaze in the in Good Standing
THE WARM-UP RING The Official News of the Jumping Committee September 2019, Volume 15, Issue 9 LETTER FROM THE CHAIR IN THIS ISSUE their phone – whatever works! – to ensure that they have all their ducks in a row for 2020 as follows: On The Show Scene • Join your National Federation Michelle C. Dunn (EC) and purchase any other Canada Second in BMO Nations’ memberships you may need to count Cup at Spruce Meadows ‘Masters’ points early in the year (provincial Royal Horse Show’s Opening hunter/jumper associations, CET Medal program registration, etc.). Weekend Showcases Canadian Double check that payment has gone Championships through and that you are a member Vote NOW for Eric Lamaze in the in good standing. • Make a decision early if you wish FEI Awards to request a change of province for BMO Introduces the Ian Millar your CET Medal Regional Final. The deadline is August 15 of each year Legacy Award with no exceptions. Thank you to our NAYC Sponsors! Fall is officially upon us, and the annual The Jumping Committee does its best to countdown to the Royal Agricultural Winter grant requests for a change of province so Get Your Jump Canada Hall of Fair is underway. With it comes the four that riders are able to compete in the region Fame Tickets! CET Medal Regional Finals which, in turn, that is most suitable for them. However, it Richard Mongeau Departs as Chief determine the riders who qualify for the cannot grant exceptions. It is clearly written Running Fox CET Medal National Final at in the Rulebook. -

The Debate on Plant and Crop Biodiversity and Biotechnology
The Debate on Plant and Crop Biodiversity and Biotechnology Klaus Ammann, [email protected] Version from December 15, 2017, 480 full text references, 117 pp. ASK-FORCE contribution No. 11 Nearly 470 references on biodiversity and Agriculture need still to be screened and selected. Contents: 1. Summary ........................................................................................................................................................................... 3 2. The needs for biodiversity – the general case ................................................................................................................ 3 3. Relationship between biodiversity and ecological parameters ..................................................................................... 5 4. A new concept of sustainability ....................................................................................................................................... 6 4.1. Revisiting the original Brundtland definition of sustainable development ...............................................................................................................7 4.2. Redefining Sustainability for Agriculture and Technology, see fig. 1 .........................................................................................................................8 5. The Issue: unnecessary stigmatization of GMOs .......................................................................................................... 12 6. Types of Biodiversity ...................................................................................................................................................... -

Multidisciplinary Approach to Genetic Variability, Inbreeding and Fertility in the Endangered Sorraia Horse Breed
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS Multidisciplinary approach to genetic variability, inbreeding and fertility in the endangered Sorraia horse breed Doutoramento em Biologia Biologia da Conservação Helena Josefina Kjöllerström Tese orientada por: Profª Doutora Maria do Mar Jácome Félix Oom Prof. Doutor Bhanu Pratap Chowdhary Documento especialmente elaborado para a obtenção do grau de doutor 2016 UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS Multidisciplinary approach to genetic variability, inbreeding and fertility in the endangered Sorraia horse breed Doutoramento em Biologia Biologia da Conservação Helena Josefina Kjöllerström Tese orientada por: Profª Doutora Maria do Mar Jácome Félix Oom Prof. Doutor Bhanu Pratap Chowdhary Júri: Presidente: ●Doutora Maria Manuela Gomes Coelho de Noronha Trancoso Vogais: ● Doutora Terje Raudsepp ● Doutora Raquel Maria Garcia dos Santos Chaves ● Doutor José António dos Santos Pereira de Matos ● Doutor Luís Lavadinho Telo da Gama ● Doutora Maria Manuela Gomes Coelho de Noronha Trancoso ● Doutora Maria do Mar Jácome Félix Oom Documento especialmente elaborado para a obtenção do grau de doutor SFRH/BD/81502/2011, cE3c UID/BIA/00329/2013, PRODER (57882)(EU)/Action 2.2.3 (2011-2013), LINK Equine Research Endowment, American Quarter Horse Foundation and USDA (#2012-67015-19632) 2016 This work was supported by FCT/MEC (Fundação para a Ciência e Tecnologia) PhD Fellowship (SFRH/BD/81502/2011), cE3c FCT Unit funding (Ref. UID/BIA/00329/2013), PRODER (Contract number 57882)(EU)/Action 2.2.3 (2011-2013) [Conservation and improvement of genetic resources, Sub-action 2.2.3.2. Animal component (Sorraia breed)], LINK Equine Research Endowment, American Quarter Horse Foundation and USDA (grant #2012-67015-19632). -

Treatment of Equine Sarcoid with the Mistletoe Extract ISCADOR® P
Pferdeklinik der Vetsuisse-Fakultät der Universität Bern (Leiter: PD. Dr. V. Gerber) Arbeit unter der Leitung von PD Dr. V. Gerber und Prof. Dr. R. Straub Treatment of Equine Sarcoid with the mistletoe extract ISCADOR® P (viscum album austriacus) - a double-blind placebo controlled study Inaugural-Dissertation zur Erlangung der Doktorwürde der Vetsuisse-Fakultät der Universität Bern vorgelegt von Ophélie Clottu von Cornaux und Neuchâtel 2008 1 Von der Vetsuisse-Fakultät der Universität Bern auf Antrag von Prof. Dr. R. Straub als Dissertation genehmigt. Bern, den Der Dekan der Vetsuisse-Fakultät der Universität Bern 2 Treatment of Equine Sarcoid with the mistletoe extract ISCADOR® P (viscum album austriacus) - a double-blind placebo controlled study Summary Background: The Equine Sarcoid (ES) is the most common skin tumour in horses. It is difficult to treat: recurrence rate is high and no single universally effective treatment for ES is known. Viscum album extracts (VAE) are used as adjunct in the treatment of some human cancers without known side effects. Hypothesis: We hypothesized that the long-term therapy with VAE (Iscador® P) is effective in Equine Sarcoids as a single treatment or as an adjunct treatment to selective surgical excision. Material and methods: Of 53 horses with ES, 42 were treated solely with VAE as monotherapy and 11 were treated after selective excision of ES. Horses were randomly assigned to the treatment group (VAE; n=32) or control group (Placebo; n=21). The horses received rising concentrations of Iscador® P extract from 0.1 mg per ml to 20 mg per ml or physiological NaCl solution 3 times a week over 105 days s.c.