Rates of Chemical Reactions: Their Measurement and Mathematical Expressions - Beltrame P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 12 Lecture Notes
Chemical Kinetics “The study of speeds of reactions and the nanoscale pathways or rearrangements by which atoms and molecules are transformed to products” Chapter 13: Chemical Kinetics: Rates of Reactions © 2008 Brooks/Cole 1 © 2008 Brooks/Cole 2 Reaction Rate Reaction Rate Combustion of Fe(s) powder: © 2008 Brooks/Cole 3 © 2008 Brooks/Cole 4 Reaction Rate Reaction Rate + Change in [reactant] or [product] per unit time. Average rate of the Cv reaction can be calculated: Time, t [Cv+] Average rate + Cresol violet (Cv ; a dye) decomposes in NaOH(aq): (s) (mol / L) (mol L-1 s-1) 0.0 5.000 x 10-5 13.2 x 10-7 10.0 3.680 x 10-5 9.70 x 10-7 20.0 2.710 x 10-5 7.20 x 10-7 30.0 1.990 x 10-5 5.30 x 10-7 40.0 1.460 x 10-5 3.82 x 10-7 + - 50.0 1.078 x 10-5 Cv (aq) + OH (aq) → CvOH(aq) 2.85 x 10-7 60.0 0.793 x 10-5 -7 + + -5 1.82 x 10 change in concentration of Cv Δ [Cv ] 80.0 0.429 x 10 rate = = 0.99 x 10-7 elapsed time Δt 100.0 0.232 x 10-5 © 2008 Brooks/Cole 5 © 2008 Brooks/Cole 6 1 Reaction Rates and Stoichiometry Reaction Rates and Stoichiometry Cv+(aq) + OH-(aq) → CvOH(aq) For any general reaction: a A + b B c C + d D Stoichiometry: The overall rate of reaction is: Loss of 1 Cv+ → Gain of 1 CvOH Rate of Cv+ loss = Rate of CvOH gain 1 Δ[A] 1 Δ[B] 1 Δ[C] 1 Δ[D] Rate = − = − = + = + Another example: a Δt b Δt c Δt d Δt 2 N2O5(g) 4 NO2(g) + O2(g) Reactants decrease with time. -

Ochem ACS Review 23 Aryl Halides
ACS Review Aryl Halides 1. Which one of the following has the weakest carbon-chlorine bond? A. I B. II C. III D. IV 2. Which compound in each of the following pairs is the most reactive to the conditions indicated? A. I and III B. I and IV C. II and III D. II and IV 3. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. 4-nitrofluorobenzene C. 2,4-dinitrofluorobenzene D. 2,4,6-trinitrofluorobenzene 4. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. p-nitrofluorobenzene C. p-fluorotoluene D. p-bromofluorobenzene 5. Which of the following is the kinetic rate equation for the addition-elimination mechanism of nucleophilic aromatic substitution? A. rate = k[aryl halide] B. rate = k[nucleophile] C. rate = k[aryl halide][nucleophile] D. rate = k[aryl halide][nucleophile] 2 6. Which of the following is not a resonance form of the intermediate in the nucleophilic addition of hydroxide ion to para -fluoronitrobenzene? A. I B. II C. III D. IV 7. Which chlorine is most susceptible to nucleophilic substitution with NaOCH 3 in methanol? A. #1 B. #2 C. #1 and #2 are equally susceptible D. no substitution is possible 8. What is the product of the following reaction? A. N,N-dimethylaniline B. para -chloro-N,N-dimethylaniline C. phenyllithium (C 6H5Li) D. meta -chloro-N,N-dimethylaniline 9. Which one of the reagents readily reacts with bromobenzene without heating? A. -

Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling
Article pubs.acs.org/JCTC Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling Calculations Matthew J. Varga and Steven D. Schwartz* Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States *S Supporting Information ABSTRACT: In this study, we develop and test a method to determine the rate of particle transfer and kinetic isotope effects in enzymatic reactions, specifically yeast alcohol dehydrogenase (YADH), from first-principles. Transition path sampling (TPS) and normal mode centroid dynamics (CMD) are used to simulate these enzymatic reactions without knowledge of their reaction coordinates and with the inclusion of quantum effects, such as zero-point energy and tunneling, on the transferring particle. Though previous studies have used TPS to calculate reaction rate constants in various model and real systems, it has not been applied to a system as large as YADH. The calculated primary H/D kinetic isotope effect agrees with previously reported experimental results, within experimental error. The kinetic isotope effects calculated with this method correspond to the kinetic isotope effect of the transfer event itself. The results reported here show that the kinetic isotope effects calculated from first-principles, purely for barrier passage, can be used to predict experimental kinetic isotope effects in enzymatic systems. ■ INTRODUCTION staging method from the Chandler group.10 Another method that uses an energy gap formalism like QCP is a hybrid Particle transfer plays a crucial role in a breadth of enzymatic ff reactions, including hydride, hydrogen atom, electron, and quantum-classical approach of Hammes-Schi er and co-work- 1 ers, which models the transferring particle nucleus as a proton-coupled electron transfer reactions. -

Chemical Oscillations 1
GENERAL I ARTICLE Chemical Oscillations 1. Basic Principles and Examples Monika Sharma and Pra'Veen Kumar A chemical reaction is usually thought of as coming to gether of reactant molecules to form products. The concen trations of initial components (reactants) decrease, and concentrations of products increase until they reach a well defined state: the equilibrium. This process is accompa nied by a decrease of the system free energy (compared at constant pressure and temperature), until it reaches a mini Monika Shanna is a JRF mum in the equilibrium. Thus, it follows from the nature of at the Protein Science and the law ofmass-action that every simple reaction approaches Engineering Unit, its equilibrium asymptotically, and the evolution of any Institute of Microbial Technology (IMTECH), physico-chemical system leads invariably to the steady Chandigarh. state ofmaximum disorder in the universe. Normally, chemi cal systems approach equilibrium in a smooth, frequently exponential relaxation. Under special circumstances, how ever, coherent behavior such as sustained oscillations are observed and the oscillations of chemical origin have been present as long as life itseH. Such reactions can be studied Praveen Kumar is using mathematical models, the Lotka-Volterra model be pursuing his PhD in ing the earliest and the simplest one. chemical dynamics at Panjab University, 1. Introduction Chandigarh. Every living system contains hundreds of chemical oscillators. Some examples are systems such as circadian clocks and rhyth mic activity -

Universal Motifs and the Diversity of Autocatalytic Systems
Universal motifs and the diversity of autocatalytic systems Alex Blokhuisa,b,c,d,1, David Lacostea, and Philippe Ngheb,1 aGulliver Laboratory, UMR CNRS 7083, Paris Sciences et Lettres University, Paris F-75231, France; bLaboratoire de Biochimie, UMR CNRS 8231, Chimie Biologie et Innovation, Ecole Superieure´ de Physique et de Chimie Industrielles de la ville de Paris, PSL University, Paris F-75231, France; cGroningen Institute for Evolutionary Life Sciences, University of Groningen, 9747 AG Groningen, The Netherlands; and dCentre for Systems Chemistry, Stratingh Institute, University of Groningen, 9747 AG Groningen, The Netherlands Edited by Peter Schuster, University of Vienna, Vienna, Austria, and approved September 1, 2020 (received for review June 30, 2020) Autocatalysis is essential for the origin of life and chemical evo- From this definition, we derive conditions to determine lution. However, the lack of a unified framework so far prevents whether a subnetwork embedded in a larger chemical network a systematic study of autocatalysis. Here, we derive, from basic can be catalytic or autocatalytic. These conditions provide a principles, general stoichiometric conditions for catalysis and auto- mathematical basis to identify minimal motifs, called autocat- catalysis in chemical reaction networks. This allows for a classifica- alytic cores. We found that cores have five fundamental cat- tion of minimal autocatalytic motifs called cores. While all known egories of motifs. They allow classification of all previously autocatalytic systems indeed contain minimal motifs, the classifica- described forms of autocatalysis, and also reveal hitherto uniden- tion also reveals hitherto unidentified motifs. We further examine tified autocatalytic schemes. We then study the kinetic condi- conditions for kinetic viability of such networks, which depends tions, which we call viability conditions, under which autocat- on the autocatalytic motifs they contain and is notably increased alytic networks can appear and be maintained on long timescales. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Universal Motifs and the Diversity of Autocatalytic Systems
Universal motifs and the diversity of autocatalytic systems Alex Blokhuisa,b,c,d,1, David Lacostea, and Philippe Ngheb,1 aGulliver Laboratory, UMR CNRS 7083, Paris Sciences et Lettres University, Paris F-75231, France; bLaboratoire de Biochimie, UMR CNRS 8231, Chimie Biologie et Innovation, Ecole Superieure´ de Physique et de Chimie Industrielles de la ville de Paris, PSL University, Paris F-75231, France; cGroningen Institute for Evolutionary Life Sciences, University of Groningen, 9747 AG Groningen, The Netherlands; and dCentre for Systems Chemistry, Stratingh Institute, University of Groningen, 9747 AG Groningen, The Netherlands Edited by Peter Schuster, University of Vienna, Vienna, Austria, and approved September 1, 2020 (received for review June 30, 2020) Autocatalysis is essential for the origin of life and chemical evo- From this definition, we derive conditions to determine lution. However, the lack of a unified framework so far prevents whether a subnetwork embedded in a larger chemical network a systematic study of autocatalysis. Here, we derive, from basic can be catalytic or autocatalytic. These conditions provide a principles, general stoichiometric conditions for catalysis and auto- mathematical basis to identify minimal motifs, called autocat- catalysis in chemical reaction networks. This allows for a classifica- alytic cores. We found that cores have five fundamental cat- tion of minimal autocatalytic motifs called cores. While all known egories of motifs. They allow classification of all previously autocatalytic systems indeed contain minimal motifs, the classifica- described forms of autocatalysis, and also reveal hitherto uniden- tion also reveals hitherto unidentified motifs. We further examine tified autocatalytic schemes. We then study the kinetic condi- conditions for kinetic viability of such networks, which depends tions, which we call viability conditions, under which autocat- on the autocatalytic motifs they contain and is notably increased alytic networks can appear and be maintained on long timescales. -

Design Principles of Autocatalytic Cycles Constrain Enzyme Kinetics and Force Low Substrate Saturation at Flux Branch Points
bioRxiv preprint doi: https://doi.org/10.1101/074641; this version posted January 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Design principles of autocatalytic cycles constrain enzyme kinetics and force low substrate saturation at flux branch points Uri Barenholz1, Dan Davidi1, Ed Reznik2,3, Yinon Bar-On1, Niv Antonovsky1, Elad Noor4, and Ron Milo1 1Department of Plant and Environmental Sciences, The Weizmann Institute of Science 2Computational Biology Program, Memorial Sloan Kettering Cancer Center 3Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center 4Institue of Molecular Systems Biology, ETH Zurich Abstract A set of chemical reactions that require a metabolite to synthesize more of that metabolite is an autocatalytic cycle. Here we show that most of the reactions in the core of central carbon metabolism are part of compact autocatalytic cycles. Such metabolic designs must meet specific conditions to support stable fluxes, hence avoiding depletion of intermediate metabolites. As such, they are subjected to constraints that may seem counter-intuitive: the enzymes of branch reactions out of the cycle must be overexpressed and the affinity of these enzymes to their substrates must be relatively weak. We use recent quantitative proteomics and fluxomics measurements to show that the above conditions hold for functioning cycles in central carbon metabolism of E.coli. This work demonstrates that the topology of a metabolic network can shape kinetic parameters of enzymes and lead to seemingly wasteful enzyme usage. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -
Microtest III Print File
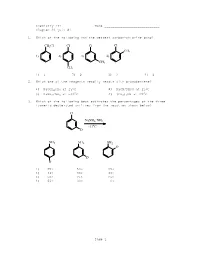
Chemistry 211 Name ____________________________ Chapter 23 Quiz #1 1. Which of the following has the weakest carbon-chlorine bond? CH2Cl Cl Cl Cl CH3 1) 2) 3) 4) CH3 CH3 1) 1 2) 2 3) 3 4) 4 2. Which one of the reagents readily reacts with bromobenzene? 1) NaOCH2CH3 at 250C 2) NaCN/DMSO at 250C 3) NaNH2/NH3 at -330C 4) (CH3)2NH at 250C 3. Which of the following best estimates the percentages of the three isomeric deuterated anilines from the reaction shown below? Cl NaNH2, NH3 -33oC D NH 2 NH 2 NH 2 D D D 1) 25% 50% 25% 2) 33% 33% 33% 3) 50% 25% 25% 4) 66% 33% 0% Page 1 4. Which of the following is (are) true concerning the intermediate in the addition-elimination mechanism of the reaction below? F OCH3 NaOCH 3 intermediate NO2 NO2 A. The intermediate is aromatic. B. The intermediate is a resonance stabilized anion. C. Electron withdrawing groups on the benzene ring stabilize the intermediate. 1) only A 2) only B 3) A and C 4) B and C 5. Carbon-14 labelled chlorobenzene is reacted with sodium amide in ammonia as shown below. Which of the following depicts the carbon-14 label in the product(s)? Cl NaNH2/NH3 -33oC Cl Cl Cl 1) 3) and (about 50% of each) Cl Cl Cl 2) 4) and (about 50% of each) 1) 1 2) 2 3) 3 4) 4 6. Which of the following reacts at the fastest rate with potassium methoxide (KOCH3) in methanol? 1) fluorobenzene 2) 4-nitrofluorobenzene 3) 2,4-dinitrofluorobenzene 4) 2,4,6-trinitrofluorobenzene Page 2 7. -

S 1 Reaction Remember That a 3˚ Alkyl Halide Will Not Undergo a S 2 Reaction the Steric Hindrance in the Transition State
SN1 Reaction Remember that a 3˚ alkyl halide will not undergo a SN2 reaction The steric hindrance in the transition state is too high If t-Butyl iodide is reacted with methanol, however, a substitution product is obtained This product does NOT proceed through a SN2 reaction First proof is that the rate for the reaction does not depend on methanol concentration Occurs through a SN1 reaction Substitution – Nucleophilic – Unimolecular (1) SN1 - In a SN1 reaction the leaving group departs BEFORE a nucleophile attacks For t-butyl iodide this generates a planar 3˚ carbocation The carbocation can then react with solvent (or nucleophile) to generate the product If solvent reacts (like the methanol as shown) the reaction is called a “solvolysis” The Energy Diagram for a SN1 Reaction therefore has an Intermediate - And is a two step reaction CH3 Potential H3C energy CH3 I OCH CH3 3 H3C CH3 CH3 H3C CH3 Reaction Coordinate Why does a SN1 Mechanism Occur? - Whenever the potential energy of a reaction is disfavored for a SN2 reaction the SN1 mechanism becomes a possibility SN2 SN1 Potential energy I OCH3 CH3 H3C CH3 CH3 H3C CH3 Reaction Coordinate Rate Characteristics The rate for a SN1 reaction is a first order reaction Rate = k [substrate] The first step is the rate determining step The nucleophile is NOT involved in the rate determining step Therefore the rate of a SN1 reaction is independent of nucleophile concentration (or nucleophile characteristics, e.g. strength) What is Important for a SN1 Reaction? The primary factor concerning the rate of -

Enthalpy and Free Energy of Reaction
KINETICS AND THERMODYNAMICS OF REACTIONS Key words: Enthalpy, entropy, Gibbs free energy, heat of reaction, energy of activation, rate of reaction, rate law, energy profiles, order and molecularity, catalysts INTRODUCTION This module offers a very preliminary detail of basic thermodynamics. Emphasis is given on the commonly used terms such as free energy of a reaction, rate of a reaction, multi-step reactions, intermediates, transition states etc., FIRST LAW OF THERMODYNAMICS. o Law of conservation of energy. States that, • Energy can be neither created nor destroyed. • Total energy of the universe remains constant. • Energy can be converted from one form to another form. eg. Combustion of octane (petrol). 2 C8H18(l) + 25 O2(g) CO2(g) + 18 H2O(g) Conversion of potential energy into thermal energy. → 16 + 10.86 KJ/mol . ESSENTIAL FACTORS FOR REACTION. For a reaction to progress. • The equilibrium must favor the products- • Thermodynamics(energy difference between reactant and product) should be favorable • Reaction rate must be fast enough to notice product formation in a reasonable period. • Kinetics( rate of reaction) ESSENTIAL TERMS OF THERMODYNAMICS. Thermodynamics. Predicts whether the reaction is thermally favorable. The energy difference between the final products and reactants are taken as the guiding principle. The equilibrium will be in favor of products when the product energy is lower. Molecule with lowered energy posses enhanced stability. Essential terms o Free energy change (∆G) – Overall free energy difference between the reactant and the product o Enthalpy (∆H) – Heat content of a system under a given pressure. o Entropy (∆S) – The energy of disorderness, not available for work in a thermodynamic process of a system.