Kramers' Theory of Reaction Kinetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 12 Lecture Notes
Chemical Kinetics “The study of speeds of reactions and the nanoscale pathways or rearrangements by which atoms and molecules are transformed to products” Chapter 13: Chemical Kinetics: Rates of Reactions © 2008 Brooks/Cole 1 © 2008 Brooks/Cole 2 Reaction Rate Reaction Rate Combustion of Fe(s) powder: © 2008 Brooks/Cole 3 © 2008 Brooks/Cole 4 Reaction Rate Reaction Rate + Change in [reactant] or [product] per unit time. Average rate of the Cv reaction can be calculated: Time, t [Cv+] Average rate + Cresol violet (Cv ; a dye) decomposes in NaOH(aq): (s) (mol / L) (mol L-1 s-1) 0.0 5.000 x 10-5 13.2 x 10-7 10.0 3.680 x 10-5 9.70 x 10-7 20.0 2.710 x 10-5 7.20 x 10-7 30.0 1.990 x 10-5 5.30 x 10-7 40.0 1.460 x 10-5 3.82 x 10-7 + - 50.0 1.078 x 10-5 Cv (aq) + OH (aq) → CvOH(aq) 2.85 x 10-7 60.0 0.793 x 10-5 -7 + + -5 1.82 x 10 change in concentration of Cv Δ [Cv ] 80.0 0.429 x 10 rate = = 0.99 x 10-7 elapsed time Δt 100.0 0.232 x 10-5 © 2008 Brooks/Cole 5 © 2008 Brooks/Cole 6 1 Reaction Rates and Stoichiometry Reaction Rates and Stoichiometry Cv+(aq) + OH-(aq) → CvOH(aq) For any general reaction: a A + b B c C + d D Stoichiometry: The overall rate of reaction is: Loss of 1 Cv+ → Gain of 1 CvOH Rate of Cv+ loss = Rate of CvOH gain 1 Δ[A] 1 Δ[B] 1 Δ[C] 1 Δ[D] Rate = − = − = + = + Another example: a Δt b Δt c Δt d Δt 2 N2O5(g) 4 NO2(g) + O2(g) Reactants decrease with time. -

Ochem ACS Review 23 Aryl Halides
ACS Review Aryl Halides 1. Which one of the following has the weakest carbon-chlorine bond? A. I B. II C. III D. IV 2. Which compound in each of the following pairs is the most reactive to the conditions indicated? A. I and III B. I and IV C. II and III D. II and IV 3. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. 4-nitrofluorobenzene C. 2,4-dinitrofluorobenzene D. 2,4,6-trinitrofluorobenzene 4. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. p-nitrofluorobenzene C. p-fluorotoluene D. p-bromofluorobenzene 5. Which of the following is the kinetic rate equation for the addition-elimination mechanism of nucleophilic aromatic substitution? A. rate = k[aryl halide] B. rate = k[nucleophile] C. rate = k[aryl halide][nucleophile] D. rate = k[aryl halide][nucleophile] 2 6. Which of the following is not a resonance form of the intermediate in the nucleophilic addition of hydroxide ion to para -fluoronitrobenzene? A. I B. II C. III D. IV 7. Which chlorine is most susceptible to nucleophilic substitution with NaOCH 3 in methanol? A. #1 B. #2 C. #1 and #2 are equally susceptible D. no substitution is possible 8. What is the product of the following reaction? A. N,N-dimethylaniline B. para -chloro-N,N-dimethylaniline C. phenyllithium (C 6H5Li) D. meta -chloro-N,N-dimethylaniline 9. Which one of the reagents readily reacts with bromobenzene without heating? A. -

Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling
Article pubs.acs.org/JCTC Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling Calculations Matthew J. Varga and Steven D. Schwartz* Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States *S Supporting Information ABSTRACT: In this study, we develop and test a method to determine the rate of particle transfer and kinetic isotope effects in enzymatic reactions, specifically yeast alcohol dehydrogenase (YADH), from first-principles. Transition path sampling (TPS) and normal mode centroid dynamics (CMD) are used to simulate these enzymatic reactions without knowledge of their reaction coordinates and with the inclusion of quantum effects, such as zero-point energy and tunneling, on the transferring particle. Though previous studies have used TPS to calculate reaction rate constants in various model and real systems, it has not been applied to a system as large as YADH. The calculated primary H/D kinetic isotope effect agrees with previously reported experimental results, within experimental error. The kinetic isotope effects calculated with this method correspond to the kinetic isotope effect of the transfer event itself. The results reported here show that the kinetic isotope effects calculated from first-principles, purely for barrier passage, can be used to predict experimental kinetic isotope effects in enzymatic systems. ■ INTRODUCTION staging method from the Chandler group.10 Another method that uses an energy gap formalism like QCP is a hybrid Particle transfer plays a crucial role in a breadth of enzymatic ff reactions, including hydride, hydrogen atom, electron, and quantum-classical approach of Hammes-Schi er and co-work- 1 ers, which models the transferring particle nucleus as a proton-coupled electron transfer reactions. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -
Microtest III Print File
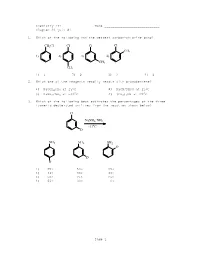
Chemistry 211 Name ____________________________ Chapter 23 Quiz #1 1. Which of the following has the weakest carbon-chlorine bond? CH2Cl Cl Cl Cl CH3 1) 2) 3) 4) CH3 CH3 1) 1 2) 2 3) 3 4) 4 2. Which one of the reagents readily reacts with bromobenzene? 1) NaOCH2CH3 at 250C 2) NaCN/DMSO at 250C 3) NaNH2/NH3 at -330C 4) (CH3)2NH at 250C 3. Which of the following best estimates the percentages of the three isomeric deuterated anilines from the reaction shown below? Cl NaNH2, NH3 -33oC D NH 2 NH 2 NH 2 D D D 1) 25% 50% 25% 2) 33% 33% 33% 3) 50% 25% 25% 4) 66% 33% 0% Page 1 4. Which of the following is (are) true concerning the intermediate in the addition-elimination mechanism of the reaction below? F OCH3 NaOCH 3 intermediate NO2 NO2 A. The intermediate is aromatic. B. The intermediate is a resonance stabilized anion. C. Electron withdrawing groups on the benzene ring stabilize the intermediate. 1) only A 2) only B 3) A and C 4) B and C 5. Carbon-14 labelled chlorobenzene is reacted with sodium amide in ammonia as shown below. Which of the following depicts the carbon-14 label in the product(s)? Cl NaNH2/NH3 -33oC Cl Cl Cl 1) 3) and (about 50% of each) Cl Cl Cl 2) 4) and (about 50% of each) 1) 1 2) 2 3) 3 4) 4 6. Which of the following reacts at the fastest rate with potassium methoxide (KOCH3) in methanol? 1) fluorobenzene 2) 4-nitrofluorobenzene 3) 2,4-dinitrofluorobenzene 4) 2,4,6-trinitrofluorobenzene Page 2 7. -

S 1 Reaction Remember That a 3˚ Alkyl Halide Will Not Undergo a S 2 Reaction the Steric Hindrance in the Transition State
SN1 Reaction Remember that a 3˚ alkyl halide will not undergo a SN2 reaction The steric hindrance in the transition state is too high If t-Butyl iodide is reacted with methanol, however, a substitution product is obtained This product does NOT proceed through a SN2 reaction First proof is that the rate for the reaction does not depend on methanol concentration Occurs through a SN1 reaction Substitution – Nucleophilic – Unimolecular (1) SN1 - In a SN1 reaction the leaving group departs BEFORE a nucleophile attacks For t-butyl iodide this generates a planar 3˚ carbocation The carbocation can then react with solvent (or nucleophile) to generate the product If solvent reacts (like the methanol as shown) the reaction is called a “solvolysis” The Energy Diagram for a SN1 Reaction therefore has an Intermediate - And is a two step reaction CH3 Potential H3C energy CH3 I OCH CH3 3 H3C CH3 CH3 H3C CH3 Reaction Coordinate Why does a SN1 Mechanism Occur? - Whenever the potential energy of a reaction is disfavored for a SN2 reaction the SN1 mechanism becomes a possibility SN2 SN1 Potential energy I OCH3 CH3 H3C CH3 CH3 H3C CH3 Reaction Coordinate Rate Characteristics The rate for a SN1 reaction is a first order reaction Rate = k [substrate] The first step is the rate determining step The nucleophile is NOT involved in the rate determining step Therefore the rate of a SN1 reaction is independent of nucleophile concentration (or nucleophile characteristics, e.g. strength) What is Important for a SN1 Reaction? The primary factor concerning the rate of -

Enthalpy and Free Energy of Reaction
KINETICS AND THERMODYNAMICS OF REACTIONS Key words: Enthalpy, entropy, Gibbs free energy, heat of reaction, energy of activation, rate of reaction, rate law, energy profiles, order and molecularity, catalysts INTRODUCTION This module offers a very preliminary detail of basic thermodynamics. Emphasis is given on the commonly used terms such as free energy of a reaction, rate of a reaction, multi-step reactions, intermediates, transition states etc., FIRST LAW OF THERMODYNAMICS. o Law of conservation of energy. States that, • Energy can be neither created nor destroyed. • Total energy of the universe remains constant. • Energy can be converted from one form to another form. eg. Combustion of octane (petrol). 2 C8H18(l) + 25 O2(g) CO2(g) + 18 H2O(g) Conversion of potential energy into thermal energy. → 16 + 10.86 KJ/mol . ESSENTIAL FACTORS FOR REACTION. For a reaction to progress. • The equilibrium must favor the products- • Thermodynamics(energy difference between reactant and product) should be favorable • Reaction rate must be fast enough to notice product formation in a reasonable period. • Kinetics( rate of reaction) ESSENTIAL TERMS OF THERMODYNAMICS. Thermodynamics. Predicts whether the reaction is thermally favorable. The energy difference between the final products and reactants are taken as the guiding principle. The equilibrium will be in favor of products when the product energy is lower. Molecule with lowered energy posses enhanced stability. Essential terms o Free energy change (∆G) – Overall free energy difference between the reactant and the product o Enthalpy (∆H) – Heat content of a system under a given pressure. o Entropy (∆S) – The energy of disorderness, not available for work in a thermodynamic process of a system. -

Kinetics and Mechanistic Investigation of Epoxide/CO2 Cycloaddition by a Synergistic Catalytic Effect of Pyrrolidinopyridinium Iodide and Zinc Halides
Kinetics and mechanistic investigation of epoxide/CO2 cycloaddition by a synergistic catalytic effect of pyrrolidinopyridinium iodide and zinc halides Abdul Rehman a, b,*, Valentine C. Eze a, M.F.M. Gunam Resula, Adam Harveya a School of Engineering, Merz Court, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK b Department of Chemical and Polymer Engineering, University of Engineering and Technology Lahore, Faisalabad Campus, Pakistan *[email protected] Abstract Formation of styrene carbonate (SC) by the cycloaddition of CO2 to styrene oxide (SO) catalysed by pyrrolidinopyridinium iodide (PPI) in combination with zinc halides (ZnCl2, ZnBr2 and ZnI2) was investigated. Complete conversion of the SO to SC was achieved in 3 h with 100% selectivity using 1:0.5 molar (PPI/ZnI2) catalyst ratio under mild reaction conditions i.e. 100 °C and 10 bar CO2 pressure. The synergistic effect of ZnI2 and PPI resulted in more than 7-fold increase in reaction rate than using PPI alone. The cycloaddition reaction demonstrated the first-order dependence with respect to the epoxide, CO2 and catalyst concentrations. Moreover, the kinetic and thermodynamic activation parameters of SC formation were determined using the Arrhenius and Eyring equations. The positive values of ΔH‡ (42.8 kJ mol–1) and ΔG‡ (102.3 kJ mol–1) revealed endergonic and chemically controlled nature of the reaction, whereas the large negative values of ΔS‡ (–159.4 J mol–1 K–1) indicate a highly ordered activated complex at the transition state. The activation energy for SC formation catalyzed by PPI alone was found to be 73.2 kJ mol–1 over a temperature range of 100–140 °C, –1 which was reduced to 46.1 kJ mol when using PPI in combination with ZnI2 as a binary catalyst. -

Chemical Kinetics, but Were Afraid to Ask…
Everything You Always Wanted to Know (or better yet, need to know) About Chemical Kinetics, But Were Afraid to Ask… Overall Reactions An overall chemical reaction indicates reactants and products in the appropriate stoichiometric quantities. Overall chemical reactions can be used to predict quantities consumed or produced and write equilibrium expressions. Consequently, overall reactions are used in thermodynamic calculations, such as energy changes and equilibrium concentrations. However, the overall reaction provides no information about the molecular process involved in transforming reactants into products, the nature and number of individual steps involved, the presence of intermediates or the role of catalysis. Consequently, overall reactions provide no information about how fast a reaction proceeds or the various factors that influence reaction rates. For example, the overall reaction for the combustion of methane is given by; CH4(g) + O2(g) ==== CO2(g) + 2 H2O(g) From which we can write and equilibrium expression, calculate free energy changes and so on, but which provide little insight into how long it may take for the reaction to occur or what species might act as catalysts. Reaction Mechanisms Mechanisms provide the individual steps that occur on the molecular level in transforming reactants into products. (Whereas the overall reaction indicates where we start and end up, the mechanism tells us how we got there). Reaction mechanisms are composed of a series of elementary processes (steps) which are indicative of the molecular encounters. Because of this, mechanisms are intimately related to chemical kinetics. For example, the first two steps in the combustion of methane are given by; CH4 + OH Æ CH3 + H2O slow CH3 + O2 Æ CH3O2 fast Interestingly, mechanisms are never proven but rather postulated and checked for consistency with observed experimental kinetic data. -

PDF (Chapter 5
~---=---5 __ Heterogeneous Catalysis 5.1 I Introduction Catalysis is a term coined by Baron J. J. Berzelius in 1835 to describe the property of substances that facilitate chemical reactions without being consumed in them. A broad definition of catalysis also allows for materials that slow the rate of a reac tion. Whereas catalysts can greatly affect the rate of a reaction, the equilibrium com position of reactants and products is still determined solely by thermodynamics. Heterogeneous catalysts are distinguished from homogeneous catalysts by the dif ferent phases present during reaction. Homogeneous catalysts are present in the same phase as reactants and products, usually liquid, while heterogeneous catalysts are present in a different phase, usually solid. The main advantage of using a hetero geneous catalyst is the relative ease of catalyst separation from the product stream that aids in the creation of continuous chemical processes. Additionally, heteroge neous catalysts are typically more tolerant ofextreme operating conditions than their homogeneous analogues. A heterogeneous catalytic reaction involves adsorption of reactants from a fluid phase onto a solid surface, surface reaction of adsorbed species, and desorption of products into the fluid phase. Clearly, the presence of a catalyst provides an alter native sequence of elementary steps to accomplish the desired chemical reaction from that in its absence. If the energy barriers of the catalytic path are much lower than the barrier(s) of the noncatalytic path, significant enhancements in the reaction rate can be realized by use of a catalyst. This concept has already been introduced in the previous chapter with regard to the CI catalyzed decomposition of ozone (Figure 4.1.2) and enzyme-catalyzed conversion of substrate (Figure 4.2.4). -

Chemical Kinetics Study of the TIME Vs RATE of Chemical Change CHEMICAL KINETICS DEALS WITH
Chemical Kinetics Study of the TIME vs RATE of Chemical Change CHEMICAL KINETICS DEALS WITH 1. How FAST {Speed like miles per hour} and 2. By what MECHANISM does a reaction happen? Part I How Fast (RATE) Does a Chemical Reaction “Go” ? What does the speed (RATE) depend upon? REACTION RATES • The change in the concentration of a reactant or product with time (M/s) for A → B ∆[A] ∆[B] Rate = − Rate = ∆t ∆t Rate disappearance = Rate of formation AFFECTS THE RATE 2A → B A disappears at twice the rate B forms Rate = - ½ ∆[A] = ∆[B] Rate disappearance ≠ Rate of formation For 2 N2O5 4 NO 2 + O 2 -7 If rate of decomposition of N 2O5 = 4.2 x 10 M/s What is the rate of APPEARANCE of (a) NO 2 ? Twice rate of decomposition = ________ M/s (b) O 2 ? ½ the rate of decomposition = _________ M/s How is the rate of the disappearance of the reactants related to the appearance of the products for CH 4(g) + 2O 2(g) →→→ CO 2(g) + 2H 2O( g) 1 ∆[CH ] 1 ∆[O ] 1 ∆[CO ] 1 ∆[H ] rate = − 4 = − 2 = 2 = 2 1 ∆t 2 ∆t 1 ∆t 2 ∆t Consider the reaction 3A --> 2B The average rate of appearance of B is given by [B]/ t. How is the average rate of appearance of B related to the average rate of disappear- ance of A? (a) -2[A]/3 t (b) 2[A]/3 t (c) -3[A]/2 t (d) [A]/ t (e) -[A]/ t Answer on Text Book’s Web Site 1.