Emerging Roles of Matricellular Proteins in Systemic Sclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Global Analysis Reveals the Complexity of the Human Glomerular Extracellular Matrix
Global analysis reveals the complexity of the human glomerular extracellular matrix Rachel Lennon,1,2 Adam Byron,1,* Jonathan D. Humphries,1 Michael J. Randles,1,2 Alex Carisey,1 Stephanie Murphy,1,2 David Knight,3 Paul E. Brenchley,2 Roy Zent,4,5 and Martin J. Humphries.1 1Wellcome Trust Centre for Cell-Matrix Research, Faculty of Life Sciences, University of Manchester, Manchester, UK; 2Faculty of Medical and Human Sciences, University of Manchester, Manchester, UK; 3Biological Mass Spectrometry Core Facility, Faculty of Life Sciences, University of Manchester, Manchester, UK; 4Division of Nephrology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA; and 5Veterans Affairs Hospital, Nashville, TN, USA. *Present address: Edinburgh Cancer Research UK Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK. Running title: Proteome of the glomerular matrix Word count: Abstract: 208, main text 2765 Corresponding author: Dr Rachel Lennon, Wellcome Trust Centre for Cell-Matrix Research, Michael Smith Building, University of Manchester, Manchester M13 9PT, UK. Phone: 0044 (0) 161 2755498. Fax: 0044 (0) 161 2755082. Email: [email protected] Abstract The glomerulus contains unique cellular and extracellular matrix (ECM) components, which are required for intact barrier function. Studies of the cellular components have helped to build understanding of glomerular disease; however, the full composition and regulation of glomerular ECM remains poorly understood. Here, we employed mass spectrometry–based proteomics of enriched ECM extracts for a global analysis of human glomerular ECM in vivo and identified a tissue-specific proteome of 144 structural and regulatory ECM proteins. This catalogue includes all previously identified glomerular components, plus many new and abundant components. -

Immunohistochemical Expression of Tenascin and Elastin In
Clinical Obstetrics, Gynecology and Reproductive Medicine Research Article ISSN: 2059-4828 Immunohistochemical expression of tenascin and elastin in women with pelvic organ prolapse and/or without stress urinary incontinence Ilias Liapis1, Panagiotis Bakas2, Pafiti-Kondi Agatha3, Matrona Frangou-Plemenou4, Charalampos Karachalios5*, Dimos Sioutis6 and Aggelos Liapis2 1Birmingham Women’s and Children’s Hospital, NHS Foundation Trust, Health Education England Midlands and East-West Midlands, Birmingham, England, United Kingdom 2Second Department of Obstetrics and Gynecology, Aretaieio University Hospital, School of Health Sciences, Medical School, National and Kapodistrian University of Athens, Athens, Attica, Greece 3Pathology Laboratory, Aretaieio University Hospital, School of Health Sciences, Medical School, National and Kapodistrian University of Athens, Athens, Attica, Greece 4Microbiology Laboratory, Aretaieio University Hospital, School of Health Sciences, Medical School, National and Kapodistrian University of Athens, Athens, Attica, Greece 5Second Department of Obstetrics and Gynecology, Aretaieio University Hospital, School of Health Sciences, Medical School, National and Kapodistrian University of Athens, Athens, Attica, Greece 6Third Department of Obstetrics and Gynecology, Attikon University Hospital, School of Health Sciences, Medical School, National and Kapodistrian University of Athens, Athens, Attica, Greece Abstract Background and aim: Pelvic organ prolapse (POP) and stress urinary incontinence (SUI) constitute entities of pelvic floor disorders and most often occur simultaneously in the same patient, adversely affecting women’s quality of life. The pathogenesis of pelvic organ prolapse and stress urinary incontinence is not fully understood. The pelvic viscera are maintained in their place thanks to interconnection of levator ani muscles, cardinal and uterosacral ligaments, and pubocervical and rectovaginal fascia. Ligaments and fascia consist mainly of connective tissue. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Matrix Gla Protein Species and Risk of Cardiovascular Events in Type 2 Diabetic Patients
Cardiovascular and Metabolic Risk ORIGINAL ARTICLE Matrix Gla Protein Species and Risk of Cardiovascular Events in Type 2 Diabetic Patients 1 3 GEERTJE W. DALMEIJER, PHD W.M. MONIQUE VERSCHUREN, PHD reduced coronary artery calcification and 1 3 YVONNE T. VAN DER SCHOUW, PHD JOLANDA M.A. BOER, PHD – 2 1 reduced risk of CVD (6 9). These effects ELKE J. MAGDELEYNS, BSC JOLINE W.J. BEULENS, PHD 2 are thought to be mediated by increased CEES VERMEER, PHD activation of MGP (10). MGP exists as various species, which OBJECTIVEd differ in their state of phosphorylation To investigate the relationship of circulating matrix Gla protein (MGP) species or carboxylation: phosphorylated, non- with incident cardiovascular disease (CVD) or coronary heart disease (CHD) in type 2 diabetic phosphorylated (desphospho-MGP patients. [dpMGP]), carboxylated (cMGP), or un- RESEARCH DESIGN AND METHODSdEPIC-NL is a prospective cohort study among carboxylated (ucMGP). Total uncarboxy- 40,011 Dutch men and women. At baseline (1993–1997), 518 participants were known to have lated MGP (t-ucMGP) is thought to be type 2 diabetes. MGP levels were measured by ELISA techniques in baseline plasma samples. The the sum of desphospho-uncarboxylated incidence of fatal and nonfatal CVD and CVD subtypesdCHD, peripheral arterial disease (PAD), MGP (dp-ucMGP) and phosphorylated- d heart failure, and stroke were obtained by linkage to national registers. Cox proportional uncarboxylated MGP (p-ucMGP) and hazard models were used to calculate hazard ratios (HRs), adjusted for sex, waist-to-hip ratio, mainly consists of p-ucMGP. physical activity, and history of CVD. Development of assays to measure RESULTSdDuring a median 11.2 years of follow-up, 160 cases of CVD were documented. -

Inactive Matrix Gla Protein Is a Novel Circulating Biomarker Predicting
www.nature.com/scientificreports OPEN Inactive matrix Gla protein is a novel circulating biomarker predicting retinal arteriolar Received: 5 June 2018 Accepted: 17 September 2018 narrowing in humans Published: xx xx xxxx Fang-Fei Wei1, Qi-Fang Huang1, Zhen-Yu Zhang1, Karel Van Keer2, Lutgarde Thijs1, Sander Trenson3, Wen-Yi Yang1, Nicholas Cauwenberghs 1, Blerim Mujaj1, Tatiana Kuznetsova1, Karel Allegaert4,5, Harry A. J. Struijker-Boudier6, Peter Verhamme7, Cees Vermeer8 & Jan A. Staessen 1,9 Active matrix Gla protein (MGP), a potent inhibitor of calcifcation in large arteries, protects against macrovascular complications. Recent studies suggested that active MGP helps maintaining the integrity of the renal and myocardial microcirculation, but its role in preserving the retinal microcirculation remains unknown. In 935 randomly recruited Flemish participants (mean age, 40.9 years; 50.3% women), we measured plasma desphospho-uncarboxylated MGP (dp–ucMGP), a marker of poor vitamin K status using an ELISA-based assay at baseline (1996–2010) and retinal microvascular diameters using IVAN software (Vasculomatic ala Nicola, version 1.1) including the central retinal arteriolar (CRAE) and venular (CRVE) equivalent and the arteriole-to-venule ratio (AVR) at follow-up (2008–2015). CRAE (P = 0.005) and AVR (P = 0.080) at follow-up decreased across tertiles of the dp–ucMGP distribution. In unadjusted models, for a doubling of dp–ucMGP at baseline, CRAE and AVR at follow-up respectively decreased by 1.40 µm (95% confdence interval [CI], 0.32 to 2.48; P = 0.011) and 0.006 (CI, 0.001 to 0.011; P = 0.016). In multivariable-adjusted models accounting for sex, baseline characteristics and follow-up duration, these estimates were −1.03 µm (CI, −1.96 to −0.11; P = 0.028) and −0.007 (CI, −0.011 to −0.002; P = 0.007). -

Effects of Dentine Matrix Extracts on Phenotype and Behaviour of Dental Pulp Progenitor Cells
The Effect of Dentine Conditioning Agents on Solubilisation of Bioactive Dentine Matrix Components and Dentine Regeneration A thesis submitted in fulfilment of the requirements of the degree of Doctor of Philosophy Cardiff University October 2015 Leili Sadaghiani Oral and Biomedical Sciences, School of Dentistry, College of Biomedical and Life Sciences, Cardiff University I II Acknowledgements At the start I would like to express my sincere appreciation to my supervisors, Prof. Alastair Sloan and Prof. Christopher Lynch for providing insight, guidance and encouragement throughout this work. As well as lending excellent support, they allowed me intellectual freedom in this project which enabled me grow as a scientist and I am very grateful for that. I would also like to thank Prof. Rachel Waddington for her valuable guidance during the project work. I also take this opportunity to express my gratitude to all members of the Mineralised Tissue Group, including the academic, technical and postdoctoral staff as well as postgraduate students. They provided immense support to this work by sharing their knowledge, expertise and above all friendship. The groups has to be congratulated for being such an efficient, cohesive and happy team. It has been a real joy and my pleasure to work with each and every member within it. Finally, I would like to thank my beloved family; my husband Saeed and my sons Soroush and Sepehr. I would not have been able to complete this journey without their love, understanding and support. I must also thank my parents for encouraging me in all of my pursuits in life. I would not be the person I am without them. -

Innate Mechanisms of Antimicrobial Defense Associated with the Avian Eggshell Megan Rose-Martel
Innate Mechanisms of Antimicrobial Defense Associated with the Avian Eggshell Megan Rose-Martel Thesis submitted to the Faculty of Graduate and Postdoctoral Studies in partial fulfillment of the requirements for the Doctorate in Philosophy degree in Cellular and Molecular Medicine Department of Cellular and Molecular Medicine Faculty of Medicine University of Ottawa © Megan Rose-Martel, Ottawa, Canada, 2015 Dedication This thesis is dedicated to the loving memory of my father. He was a man in constant pursuit of knowledge. I was constantly reminded of how proud he was of my accomplishments, both personal and scientific. He read every article I published, every poster I created and inquired constantly about the research I was conducting, wanting to know every detail. His unwavering support and encouragement was invaluable to the completion of this thesis. I am extremely saddened that he is not here with us to see the completion of this chapter of my life which would have never been possible without him. I also dedicate this thesis to my loving husband, my wonderful mother and my darling daughter for their endless love, patience and support that they show towards me every day of my life. They have helped me through the most difficult times by listening to me, making me laugh and always having faith that I could make it. They are and will always be a constant source of comfort, love and happiness. ii Acknowledgments I would like to express my sincere gratitude to my supervisor and mentor, Dr. Maxwell Hincke, for the opportunity to work in his laboratory. His encouragement, guidance and motivation throughout my Ph.D. -

2598 Biomineralization of Bone: a Fresh View of the Roles of Non-Collagen
[Frontiers in Bioscience 16, 2598-2621, June 1, 2011] Biomineralization of bone: a fresh view of the roles of non-collagenous proteins Jeffrey Paul Gorski1 1Center of Excellence in the Study of Musculoskeletal and Dental Tissues and Dept. of Oral Biology, Sch. Of Dentistry, Univ. of Missouri-Kansas City, Kansas City, MO 64108 TABLE OF CONTENTS 1. Abstract 2. Introduction 3. Proposed mechanisms of mineral nucleation in bone 3.1. Biomineralization Foci 3.2. Calcospherulites 3.3. Matrix vesicles 4. The role of individual non-collagenous proteins 4.1. Bone Sialoprotein 4.2. Noggin 4.3. Chordin 4.4. Osteopontin 4.5. Osteopontin, bone sialoprotein, and DMP1 form individual complexes with MMPs 4.6. Bone acidic glycoprotein-75 4.7. Dentin matrix protein1 4.8. Osteocalcin 4.9. Fetuin (alpha2HS-glycoprotein) 4.10. Periostin 4.11. Tissue nonspecific alkaline phosphatase 4.12. Phospho 1 phosphatase 4.13. Ectonucleotide pyrophosphatase/phosphodiesterase 4.14. Biological effects of hydroxyapatite on bone matrix proteins 4.15. Sclerostin 4.16. Tenascin C 4.17. Phosphate-regulating neutral endopeptidase (PHEX) 4.18. Matrix extracellular phosphoglycoprotein (MEPE, OF45) 4.19. Functional importance of proteolysis in activation of transglutaminase and PCOLCE 4.20. Neutral proteases in bone 5. Summary and Perspective 6. Acknowledgements 7. References 1. ABSTRACT The impact of genetics has dramatically affected nteractions which act in positive and negative ways to our understanding of the functions of non-collagenous regulate the process of bone mineralization. Several new proteins. Specifically, mutations and knockouts have examples from the author’s laboratory are provided defined their cellular spectrum of actions. -

Dentine Sialophosphoprotein Signal in Dentineogenesis and Dentine Regeneration M.M
EuropeanMM Liu et Cells al. and Materials Vol. 42 2021 (pages 43-62) DOI: 10.22203/eCM.v042a04 DSPP signalling in dentine ISSN formation 1473-2262 DENTINE SIALOPHOSPHOPROTEIN SIGNAL IN DENTINEOGENESIS AND DENTINE REGENERATION M.M. Liu1,2, W.T. Li1,3, X.M. Xia1,4, F. Wang5, M. MacDougall6 and S. Chen1 1 Department of Developmental Dentistry, School of Dentistry, the University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA 2 Department of Endodontics, School of Stomatology, Tongji University, Shanghai, 200072, China 3 Department of Pathology, Weifang Medical University, Weifang, 261053, China 4 Department of Obstetrics and Gynaecology, Second Xiangya Hospital, Central South University Changsha, 410011, China 5 Department of Anatomy, Fujian Medical University, Fuzhou, 350122, China 6 UBC Faculty of Dentistry, University of British Columbia, Vancouver, BC, V6T 1Z3, Canada Abstract Dentineogenesis starts on odontoblasts, which synthesise and secrete non-collagenous proteins (NCPs) and collagen. When dentine is injured, dental pulp progenitors/mesenchymal stem cells (MSCs) can migrate to the injured area, differentiate into odontoblasts and facilitate formation of reactionary dentine. Dental pulp progenitor cell/MSC differentiation is controlled at given niches. Among dental NCPs, dentine sialophosphoprotein (DSPP) is a member of the small integrin-binding ligand N-linked glycoprotein (SIBLING) family, whose members share common biochemical characteristics such as an Arg-Gly-Asp (RGD) motif. DSPP expression is cell- and tissue-specific and highly seen in odontoblasts and dentine. DSPP mutations cause hereditary dentine diseases. DSPP is catalysed into dentine glycoprotein (DGP)/sialoprotein (DSP) and phosphoprotein (DPP) by proteolysis. DSP is further processed towards active molecules. -

Tenascins in Stem Cell Niches
MATBIO-01031; No of Pages 12 Matrix Biology xxx (2014) xxx–xxx Contents lists available at ScienceDirect Matrix Biology journal homepage: www.elsevier.com/locate/matbio Tenascins in stem cell niches Ruth Chiquet-Ehrismann a,b,⁎,1,GertraudOrendc,d,e,f,1, Matthias Chiquet g,1, Richard P. Tucker h,1, Kim S. Midwood i,1 a Friedrich Miescher Institute of Biomedical Research, Novartis Research Foundation, Maulbeerstrasse 66 CH-4058 Basel, Switzerland b Faculty of Sciences, University of Basel, Switzerland c Inserm U1109, The Microenvironmental Niche in Tumorigenesis and Targeted Therapy (MNT3) Team, 3 av. Molière, 67200 Strasbourg, France d Université de Strasbourg, 67000 Strasbourg, France e LabEx Medalis, Université de Strasbourg, 67000 Strasbourg, France f Fédération de Médecine Translationnelle de Strasbourg (FMTS), 67000 Strasbourg, France g Department of Orthodontics and Dentofacial Orthopedics, University of Bern, Freiburgstrasse 7, CH-3010 Bern, Switzerland h Department of Cell Biology and Human Anatomy, University of California Davis, Davis, CA 95616, USA i The Kennedy Institute of Rheumatology, University of Oxford, Roosevelt Drive, Headington, Oxford OX3 7FY, United Kingdom article info abstract Available online xxxx Tenascins are extracellular matrix proteins with distinct spatial and temporal expression during development, tissue homeostasis and disease. Based on their expression patterns and knockout phenotypes an important Keywords: role of tenascins in tissue formation, cell adhesion modulation, regulation of proliferation and -

Deciphering the Relevance of Bone ECM Signaling
cells Review Deciphering the Relevance of Bone ECM Signaling Natividad Alcorta-Sevillano y, Iratxe Macías y, Arantza Infante * and Clara I. Rodríguez * Stem Cells and Cell Therapy Laboratory, Biocruces Bizkaia Health Research Institute, Cruces University Hospital, Plaza de Cruces S/N, Barakaldo, 48903 Bizkaia, Spain; [email protected] (N.A.-S.); [email protected] (I.M.) * Correspondence: [email protected] (A.I.); [email protected] (C.I.R.) These authors contributed equally. y Received: 20 October 2020; Accepted: 7 December 2020; Published: 7 December 2020 Abstract: Bone mineral density, a bone matrix parameter frequently used to predict fracture risk, is not the only one to affect bone fragility. Other factors, including the extracellular matrix (ECM) composition and microarchitecture, are of paramount relevance in this process. The bone ECM is a noncellular three-dimensional structure secreted by cells into the extracellular space, which comprises inorganic and organic compounds. The main inorganic components of the ECM are calcium-deficient apatite and trace elements, while the organic ECM consists of collagen type I and noncollagenous proteins. Bone ECM dynamically interacts with osteoblasts and osteoclasts to regulate the formation of new bone during regeneration. Thus, the composition and structure of inorganic and organic bone matrix may directly affect bone quality. Moreover, proteins that compose ECM, beyond their structural role have other crucial biological functions, thanks to their ability to bind multiple interacting partners like other ECM proteins, growth factors, signal receptors and adhesion molecules. Thus, ECM proteins provide a complex network of biochemical and physiological signals. Herein, we summarize different ECM factors that are essential to bone strength besides, discussing how these parameters are altered in pathological conditions related with bone fragility. -
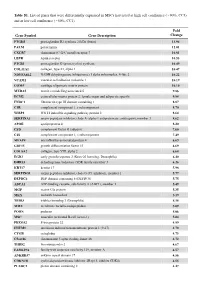
Table S1. List of Genes That Were Differentially Expressed in Mscs Harvested at High Cell Confluence (~90%, CC3) and at Low Cell Confluence (~50%, CC1)
Table S1. List of genes that were differentially expressed in MSCs harvested at high cell confluence (~90%, CC3) and at low cell confluence (~50%, CC1). Fold Gene Symbol Gene Description Change PTGDS prostaglandin D2 synthase 21kDa (brain) 13.94 PALM paralemmin 12.01 CXCR7 chemokine (C-X-C motif) receptor 7 10.81 LEPR leptin receptor 10.53 PTGIS prostaglandin I2 (prostacyclin) synthase 10.49 COL11A1 collagen, type XI, alpha 1 10.47 NDUFA4L2 NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like 2 10.22 VCAM1 vascular cell adhesion molecule 1 10.19 COMP cartilage oligomeric matrix protein 10.10 MXRA5 matrix-remodelling associated 5 9.86 ECM2 extracellular matrix protein 2, female organ and adipocyte specific 9.84 FNDC1 fibronectin type III domain containing 1 8.87 C1R complement component 1, r subcomponent 8.70 WISP2 WNT1 inducible signaling pathway protein 2 8.64 SERPINA3 serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3 8.62 APOE apolipoprotein E 8.20 CFD complement factor D (adipsin) 7.60 C1S complement component 1, s subcomponent 7.49 MFAP4 microfibrillar-associated protein 4 6.69 GDF15 growth differentiation factor 15 6.69 COL8A2 collagen, type VIII, alpha 2 6.68 EGR2 early growth response 2 (Krox-20 homolog, Drosophila) 6.40 DHRS3 dehydrogenase/reductase (SDR family) member 3 6.26 KRT17 keratin 17 5.98 SERPING1 serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 5.77 DEPDC6 DEP domain containing 6 (DEPDC6) 5.75 ABCA1 ATP-binding cassette, sub-family A (ABC1), member 1 5.49 MGP matrix