A Molecular Dynamics Study Into Annexin A1 Induced Membrane Binding and Aggregation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Gene Expression Signature for Biliary Atresia and a Role for Interleukin-8
SUPPLEMENTARY “PATIENTS AND METHODS” “Gene expression signature for biliary atresia and a role for Interleukin-8 in pathogenesis of experimental disease” Bessho K, et al. PATIENTS AND METHODS Patients. Liver biopsies, serum samples and clinical data were obtained from infants with cholestasis enrolled into a prospective study (ClinicalTrials.gov Identifier: NCT00061828) of the NIDDK-funded Childhood Liver Disease Research and Education Network (www.childrennetwork.org) or from infants evaluated at Cincinnati Children’s Hospital Medical Center. For subjects with biliary atresia (BA), liver biopsies were obtained from 64 infants during the preoperative workup or at the time of intraoperative cholangiogram, with ages ranging from 22-169 days after birth (Supplementary Table 7). For subjects with intrahepatic cholestasis (serving as diseased controls, and referred to as non- BA), liver biopsy samples were obtained percutaneously or intraoperatively from 14 infants at the time of diagnostic evaluation, with ages ranging from 19-189 days. Their diagnosis were Alagille syndrome (N=1), multidrug resistance protein-3 deficiency (N=2), alpha-1-antitrypsin deficiency (N=2) and cholestasis with unknown etiology (N=9) (Supplementary Table 7). Representative liver biopsy photomicrographs are shown in Supplementary Figure 6A-D. A third group of normal controls (NC) consisted of liver biopsy samples obtained from 7 deceased-donor children aged 22-42 months as described previously (1). This group serves as a reference cohort, with the median levels of gene expression used to normalize gene expression across all patients in the BA and non-BA groups. This greatly facilitates the visual identification of key differences in gene expression levels between BA and non-BA groups. -

Table SII. Significantly Differentially Expressed Mrnas of GSE23558 Data Series with the Criteria of Adjusted P<0.05 And
Table SII. Significantly differentially expressed mRNAs of GSE23558 data series with the criteria of adjusted P<0.05 and logFC>1.5. Probe ID Adjusted P-value logFC Gene symbol Gene title A_23_P157793 1.52x10-5 6.91 CA9 carbonic anhydrase 9 A_23_P161698 1.14x10-4 5.86 MMP3 matrix metallopeptidase 3 A_23_P25150 1.49x10-9 5.67 HOXC9 homeobox C9 A_23_P13094 3.26x10-4 5.56 MMP10 matrix metallopeptidase 10 A_23_P48570 2.36x10-5 5.48 DHRS2 dehydrogenase A_23_P125278 3.03x10-3 5.40 CXCL11 C-X-C motif chemokine ligand 11 A_23_P321501 1.63x10-5 5.38 DHRS2 dehydrogenase A_23_P431388 2.27x10-6 5.33 SPOCD1 SPOC domain containing 1 A_24_P20607 5.13x10-4 5.32 CXCL11 C-X-C motif chemokine ligand 11 A_24_P11061 3.70x10-3 5.30 CSAG1 chondrosarcoma associated gene 1 A_23_P87700 1.03x10-4 5.25 MFAP5 microfibrillar associated protein 5 A_23_P150979 1.81x10-2 5.25 MUCL1 mucin like 1 A_23_P1691 2.71x10-8 5.12 MMP1 matrix metallopeptidase 1 A_23_P350005 2.53x10-4 5.12 TRIML2 tripartite motif family like 2 A_24_P303091 1.23x10-3 4.99 CXCL10 C-X-C motif chemokine ligand 10 A_24_P923612 1.60x10-5 4.95 PTHLH parathyroid hormone like hormone A_23_P7313 6.03x10-5 4.94 SPP1 secreted phosphoprotein 1 A_23_P122924 2.45x10-8 4.93 INHBA inhibin A subunit A_32_P155460 6.56x10-3 4.91 PICSAR P38 inhibited cutaneous squamous cell carcinoma associated lincRNA A_24_P686965 8.75x10-7 4.82 SH2D5 SH2 domain containing 5 A_23_P105475 7.74x10-3 4.70 SLCO1B3 solute carrier organic anion transporter family member 1B3 A_24_P85099 4.82x10-5 4.67 HMGA2 high mobility group AT-hook 2 A_24_P101651 -

Human/Mouse Annexin A13 Antibody Antigen Affinity-Purified Polyclonal Goat Igg Catalog Number: AF4149
Human/Mouse Annexin A13 Antibody Antigen Affinity-purified Polyclonal Goat IgG Catalog Number: AF4149 DESCRIPTION Species Reactivity Human/Mouse Specificity Detects endogenous human and mouse Annexin A13 in Western blots. In Western blots, this antibody did not crossreact with recombinant human Annexin A1, A2, A3, A4, A5, A6, A7, A8, A9, A10, or A11. Source Polyclonal Goat IgG Purification Antigen Affinitypurified Immunogen E. coliderived recombinant human Annexin A13 Met1His316 Accession # P27216 Formulation Lyophilized from a 0.2 μm filtered solution in PBS with Trehalose. See Certificate of Analysis for details. *Small pack size (SP) is supplied either lyophilized or as a 0.2 μm filtered solution in PBS. APPLICATIONS Please Note: Optimal dilutions should be determined by each laboratory for each application. General Protocols are available in the Technical Information section on our website. Recommended Sample Concentration Western Blot 1 µg/mL See Below DATA Western Blot Detection of Human/Mouse Annexin A13 by Western Blot. Western blot shows lysates of human and mouse small intestine tissue. PVDF membrane was probed with 1 µg/mL of Human/Mouse Annexin A13 Antigen Affinitypurified Polyclonal Antibody (Catalog # AF4149) followed by HRPconjugated AntiGoat IgG Secondary Antibody (Catalog # HAF017). A specific band was detected for Annexin A13 at approximately 36 kDa (as indicated). This experiment was conducted under reducing conditions and using Immunoblot Buffer Group 1. PREPARATION AND STORAGE Reconstitution Reconstitute at 0.2 mg/mL in sterile PBS. Shipping The product is shipped at ambient temperature. Upon receipt, store it immediately at the temperature recommended below. -

Time Course of Renal Transcriptomics After Subchronic Exposure to Ochratoxin a in Fisher Rats
toxins Article Time Course of Renal Transcriptomics after Subchronic Exposure to Ochratoxin A in Fisher Rats Laura Pastor 1,2,†,‡, Ariane Vettorazzi 1,2,*,† , Elizabeth Guruceaga 2,3 and Adela López de Cerain 1,2,† 1 Department of Pharmacology and Toxicology, Faculty of Pharmacy and Nutrition, University of Navarra, CIFA Building, c/Irunlarrea 1, E-31008 Pamplona, Spain; [email protected] (L.P.); [email protected] (A.L.d.C.) 2 IdiSNA, Navarra Institute for Health Research, E-31008 Pamplona, Spain; [email protected] 3 Bioinformatics Platform, Center for Applied Medical Research (CIMA), University of Navarra, E-31008 Pamplona, Spain * Correspondence: [email protected]; Tel.: +34-948425600 † These authors contributed equally to this work. ‡ Current address: General Mills, 31570 San Adrián, Navarra, Spain. Abstract: The mycotoxin ochratoxin A (OTA) is a potent nephrocarcinogen, mainly in male rats. The aim of this study was to determine the time course of gene expression (GeneChip® Rat Gene 2.0 ST Array, Affymetrix) in kidney samples from male and female F344 rats, treated daily (p.o) with 0.50 mg/kg b.w. (body weight) of OTA for 7 or 21 days, and evaluate if there were differences between both sexes. After OTA treatment, there was an evolution of gene expression in the kidney over time, with more differentially expressed genes (DEG) at 21 days. The gene expression time course was different between sexes with respect to the number of DEG and the direction of expression (up or down): the female response was progressive and consistent over time, whereas males had a different early response with more DEG, most of them up-regulated. -

Annexin A13 (ANXA13) (NM 004306) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC223936 Annexin A13 (ANXA13) (NM_004306) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Annexin A13 (ANXA13) (NM_004306) Human Tagged ORF Clone Tag: Myc-DDK Symbol: ANXA13 Synonyms: ANX13; ISA Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC223936 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGGCAATCGTCATGCTAAAGCGAGCAGTCCTCAGGGTTTTGATGTGGATCGAGATGCCAAAAAGCTGA ACAAAGCCTGCAAAGGAATGGGGACCAATGAAGCAGCCATCATTGAAATCTTATCGGGCAGGACATCAGA TGAGAGGCAACAAATCAAGCAAAAGTACAAGGCAACGTACGGCAAGGAGCTGGAGGAAGTACTCAAGAGT GAGCTGAGTGGAAACTTCGAGAAGACAGCGTTGGCCCTTCTGGACCATCCCAGCGAGTACGCCGCCCGGC AGCTGCAGAAGGCTATGAAGGGTCTGGGCACAGATGAGTCCGTCCTCATTGAGGTCCTGTGCACGAGGAC CAATAAGGAAATCATCGCCATTAAAGAGGCCTACCAAAGGCTATTTGATAGGAGCCTCGAATCAGATGTC AAAGGTGATACAAGTGGAAACCTAAAAAAAATCCTGGTGTCTCTGCTGCAGGCTAATCGCAATGAAGGAG ATGACGTGGACAAAGATCTAGCTGGTCAGGATGCCAAAGATCTGTATGATGCAGGGGAAGGCCGCTGGGG CACTGATGAGCTTGCGTTCAATGAAGTCCTGGCCAAGAGGAGCTACAAGCAGTTACGAGCCACCTTTCAA GCCTATCAAATTCTCATTGGCAAAGACATAGAAGAAGCCATTGAAGAAGAAACATCAGGCGACTTGCAGA AGGCCTATTTAACTCTCGTGAGATGTGCCCAGGATTGTGAGGACTATTTTGCTGAACGTCTGTACAAGTC GATGAAGGGTGCGGGGACCGATGAGGAGACGTTGATTCGCATAATCGTGACCAGGGCCGAGGTGGACCTT CAGGGGATCAAAGCAAAGTTCCAAGAGAAGTATCAGAAGTCTCTCTCTGACATGGTTCGCTCAGATACCT -

A Comparative Quantitative LC-MS/MS Profiling Analysis Of
proteomes Article A Comparative Quantitative LC-MS/MS Profiling Analysis of Human Pancreatic Adenocarcinoma, Adjacent-Normal Tissue, and Patient-Derived Tumour Xenografts Orla Coleman 1,* , Michael Henry 1, Fiona O’Neill 1 , Sandra Roche 1, Niall Swan 2, Lorraine Boyle 2, Jean Murphy 2, Justine Meiller 1, Neil T. Conlon 1, Justin Geoghegan 2, Kevin C. Conlon 2,3, Vincent Lynch 1,2, Ninfa L. Straubinger 4, Robert M. Straubinger 4, Gerard McVey 2,5, Michael Moriarty 1,5,†, Paula Meleady 1,† and Martin Clynes 1,† 1 National Institute for Cellular Biotechnology, Dublin City University, Glasnevin, Dublin 9, Ireland; [email protected] (M.H.); fi[email protected] (F.O.); [email protected] (S.R.); [email protected] (J.M.); [email protected] (N.T.C.); [email protected] (V.L.); [email protected] (M.M.); [email protected] (P.M.); [email protected] (M.C.) 2 St. Vincent’s University Hospital, Dublin 4, Ireland; [email protected] (N.S.); [email protected] (L.B.); [email protected] (J.M.); [email protected] (J.G.); [email protected] (K.C.C.); [email protected] (G.M.) 3 Trinity College Dublin, College Green, Dublin 2, Ireland 4 Department of Pharmaceutical Sciences, University at Buffalo, State University of New York, Buffalo, NY 14214, USA; [email protected] (N.L.S.); [email protected] (R.M.S.) 5 St. Luke’s Hospital, Highfield Road, Rathgar, Dublin 6, Ireland * Correspondence: [email protected]; Tel.: +353-01-700-5700 † These authors contributed equally to this work. -

Annexin A5 As a Marker for Hepatocellular Carcinoma in Cirrhotic Hepatitis C Virus Patients Waleed Mohamed Serag1* and Basem Eysa Elsayed2
Serag and Elsayed Egyptian Liver Journal (2021) 11:32 https://doi.org/10.1186/s43066-021-00101-y Egyptian Liver Journal ORIGINAL RESEARCH ARTICLE Open Access Annexin A5 as a marker for hepatocellular carcinoma in cirrhotic hepatitis C virus patients Waleed Mohamed Serag1* and Basem Eysa Elsayed2 Abstract Background: The tumorigenesis and development in a variety of cancers is reportedly encouraged via Annexin A5. The levels of Annexin A5 were tested in patients with or without HCC who were affected by liver cirrhosis. The objective of our study was to detect Annexin A5 levels in such patients in order to assess their function as an HCC marker. The longitudinal study comprised 91 cirrhotic HCV patients with and without HCC, and 20 healthy volunteers in the control group approved by the National Hepatology and Tropical Medicine Research Institute (NHTMRI) between March 2017 and August 2018.The serum levels Annexin A5 were found in all groups with ELISA. ANOVA, Mann-Whitney, and χ2 tests had been applied. Results: High scales of Annexin A5 (3.89 + 0.85) were recorded for cirrhosis with HCC, either than cirrhotic patients without HCC (3.06 ± 0.88) (P = 0.041), and either than the control group (0.54 ± 0.11) (P < 0.001). Conclusion: In HCV cirrhotic patients with and without HCC, AnxA5 can be used as HCC marker. Keywords: AnxA5 level, HCV, HCC, Cirrhosis Background of the 36 kDa molecular weight annexin family which is One of the world’s most common cancers is hepatocel- based on the Ca2+ membrane binding [4]. Inhibition of lular carcinomas (HCC). -

Transcriptome-Based Analysis of Kidney Gene Expression Changes Associated with Diabetes in OVE26 Mice, in the Presence and Absence of Losartan Treatment
Transcriptome-Based Analysis of Kidney Gene Expression Changes Associated with Diabetes in OVE26 Mice, in the Presence and Absence of Losartan Treatment Radko Komers1,2, Bei Xu1,2,YiFu1,2, Aaron McClelland4, Phillip Kantharidis4, Amit Mittal3, Herbert T. Cohen3, David M. Cohen1,2* 1 Division of Nephrology and Hypertension, Department of Medicine, Oregon Health & Science University, Portland, Oregon, 2 Portland V. A. Medical Center, Portland, Oregon, United States of America, 3 Nephrology Section, Boston University School of Medicine, Boston, Massachusetts, United States of America, 4 JDRF Danielle Alberti Memorial Centre for Diabetes Complications, Diabetes Division, Baker IDI Heart and Diabetes Institute, Melbourne, Australia Abstract Diabetes is among the most common causes of end-stage renal disease, although its pathophysiology is incompletely understood. We performed next-generation sequencing-based transcriptome analysis of renal gene expression changes in the OVE26 murine model of diabetes (age 15 weeks), relative to non-diabetic control, in the presence and absence of short- term (seven-day) treatment with the angiotensin receptor blocker, losartan (n = 3–6 biological replicates per condition). We detected 1438 statistically significant changes in gene expression across conditions. Of the 638 genes dysregulated in diabetes relative to the non-diabetic state, .70% were downregulation events. Unbiased functional annotation of genes up- and down-regulated by diabetes strongly associated (p,161028) with terms for oxidative stress and for endoplasmic reticulum stress/protein folding. Most of the individual gene products up- or down-regulated with diabetes were unaffected by losartan treatment; however, of the gene products dysregulated in diabetes and influenced by losartan treatment, the vast majority of changes were in the direction of amelioration rather than exacerbation of the diabetic dysregulation. -

Whole Rat DNA Array Survey for Candidate Genes Related To
EXPERIMENTAL AND THERAPEUTIC MeDICINE 2: 201-212, 2011 Whole rat DNA array survey for candidate genes related to hypertension in kidneys from three spontaneously hypertensive rat substrains at two stages of age and with hypotensive induction caused by hydralazine hydrochloride KOSHO Kinoshita, MOHAMMAD SAID ASHeNAGAR, MASAKI TABUCHI and HIDeAKI HIGASHINO Department of Pharmacology, Kinki University School of Medicine, Osaka 589-8511, Japan Received October 1, 2010; Accepted December 28, 2010 DOI: 10.3892/etm.2011.193 Abstract. Clarification of the genetic nature and more effective not markedly affect quality of life when it is moderate and care for hypertension are required, given the high incidences not serious. However, when left untreated it leads to life- of cardiovascular and cerebrovascular mortality. Thus, we threatening diseases associated with atherosclerosis, including surveyed candidate genes for hypertension with rat whole gene myocardial infarction, renal failure and strokes (1-3). In Japan, DNA microarrays using three novel methods. Gene expression where general medical care should be sufficient to treat analyses were conducted as follows: Method 1, three types of hypertension, more than 300,000 patients per year die from spontaneously hypertensive rat (SHR) substrains, SHR, stroke- diseases that are related to hypertension (4,5). Clarification of prone SHR (SHRSP) and malignant type of SHRSP (M-SHRSP) the genetic nature and more effective care for hypertension were used and compared to normotensive Wistar Kyoto rats; are required. Method 2, the expressed genes between rats of different ages There are a number of methods for investigating the genetic were compared for different blood pressures; and Method 3, nature of hypertension. -
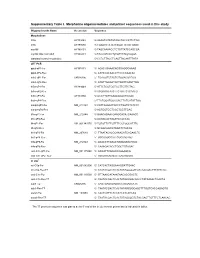
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Oncorhynchus Mykiss) After Yersinia Ruckeri Infection Gokhlesh Kumar1* , Karin Hummel2, Ebrahim Razzazi‑Fazeli2 and Mansour El‑Matbouli1
Kumar et al. Vet Res (2019) 50:54 https://doi.org/10.1186/s13567-019-0673-8 RESEARCH ARTICLE Open Access Modulation of posterior intestinal mucosal proteome in rainbow trout (Oncorhynchus mykiss) after Yersinia ruckeri infection Gokhlesh Kumar1* , Karin Hummel2, Ebrahim Razzazi‑Fazeli2 and Mansour El‑Matbouli1 Abstract Yersinia ruckeri is the causative agent of enteric redmouth disease in salmonids. In fsh, the intestine represents an important site of nutrient uptake, host–pathogen interactions, and defense. The posterior intestine can be infamed, reddened, and flled with an opaque, yellowish fuid during Y. ruckeri infection. Herein, we report an investigation on the proteome alteration in the posterior intestinal mucosa of rainbow trout (Oncorhynchus mykiss) after exposure to Y. ruckeri. The intestinal mucosal proteins were identifed and quantifed by a shotgun proteomic approach by applying data‑independent quantifcation with sequential windowed acquisition of all theoretical mass spectra (SWATH). A total of 437 proteins were found to be diferentially up‑ or downregulated in the posterior intestine. Gene ontology of upregulated proteins pointed to their involvement into exopeptidase, endopeptidase, and hydrolase activities, while the downregulated proteins were involved in lipid metabolism, actin binding, and translation processes. Additionally, upregulated proteins were predicted to be involved in lysosome, oxidative phosphorylation, and metabolic pathways, while downregulated proteins were implicated in focal adhesion, regulation of actin cytoskeleton, protein digestion and absorption pathways. This study showed that Y. ruckeri infection can alter protein abundance involved in serine‑ type carboxypeptidase, cysteine and aspartic‑type endopeptidases, metallopeptidases, antioxidant defense, calcium ion binding, glycolytic and carbohydrate metabolic processes in the proteome of the intestinal mucosa of rainbow trout.