A New Rhesus Macaque Assembly and Annotation for Next-Generation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Table SI. Primer List of Genes Used for Reverse Transcription‑Quantitative PCR Validation
Table SI. Primer list of genes used for reverse transcription‑quantitative PCR validation. Genes Forward (5'‑3') Reverse (5'‑3') Length COL1A1 AGTGGTTTGGATGGTGCCAA GCACCATCATTTCCACGAGC 170 COL6A1 CCCCTCCCCACTCATCACTA CGAATCAGGTTGGTCGGGAA 65 COL2A1 GGTCCTGCAGGTGAACCC CTCTGTCTCCTTGCTTGCCA 181 DCT CTACGAAACCAGGATGACCGT ACCATCATTGGTTTGCCTTTCA 192 PDE4D ATTGCCCACGATAGCTGCTC GCAGATGTGCCATTGTCCAC 181 RP11‑428C19.4 ACGCTAGAAACAGTGGTGCG AATCCCCGGAAAGATCCAGC 179 GPC‑AS2 TCTCAACTCCCCTCCTTCGAG TTACATTTCCCGGCCCATCTC 151 XLOC_110310 AGTGGTAGGGCAAGTCCTCT CGTGGTGGGATTCAAAGGGA 187 COL1A1, collagen type I alpha 1; COL6A1, collagen type VI, alpha 1; COL2A1, collagen type II alpha 1; DCT, dopachrome tautomerase; PDE4D, phosphodiesterase 4D cAMP‑specific. Table SII. The differentially expressed mRNAs in the ParoAF_Control group. Gene ID logFC P‑Value Symbol Description ENSG00000165480 ‑6.4838 8.32E‑12 SKA3 Spindle and kinetochore associated complex subunit 3 ENSG00000165424 ‑6.43924 0.002056 ZCCHC24 Zinc finger, CCHC domain containing 24 ENSG00000182836 ‑6.20215 0.000817 PLCXD3 Phosphatidylinositol‑specific phospholipase C, X domain containing 3 ENSG00000174358 ‑5.79775 0.029093 SLC6A19 Solute carrier family 6 (neutral amino acid transporter), member 19 ENSG00000168916 ‑5.761 0.004046 ZNF608 Zinc finger protein 608 ENSG00000134343 ‑5.56371 0.01356 ANO3 Anoctamin 3 ENSG00000110400 ‑5.48194 0.004123 PVRL1 Poliovirus receptor‑related 1 (herpesvirus entry mediator C) ENSG00000124882 ‑5.45849 0.022164 EREG Epiregulin ENSG00000113448 ‑5.41752 0.000577 PDE4D Phosphodiesterase -

DNA Ism1 823
DNA ism1 823 ATGTTGCGACTGGCAGCGGAGCTTCTGCTTCTCCTGGGACTGCTCCTCCTCACCCTGCAC 882 uninjected_emb1 ATGTTGCGACTGGCAGCGGAGCTTCTGCTTCTCCTGGGACTGCTCCTCCTCACCCTGCAC ism1_T3_emb1.1 ATGTTGCGACTGGCAGCGGAGCTTCTGCTTCTCCTGGGACTGCTCCTCCTCACCCTGCAC ism1_T3_emb1.2 ATGTTGCGACTGGCAGCGGAGCTTCTGCTTCTCCTGGGACTGCTCCTCCTCACCCTGCAC ism1_T3_emb1.3 ATGTTGCGACTGGCAGCGGAGCTTCTGCTTCTCCTGGGACTGCTCCTCCTCCTCATACAG ism1_T3_emb1.4 ATGTTGCGACTGGCAGCGGAGCTTCTGCTTCTCCTGGGACTGCTCCTCCTCCTCATACAG ism1 883 ATCACTGTGCTCCGAGGCAGCCCCGATAGCTCCTCCAACTCCAGCCACAGCCTCATACAG 942 uninjected_emb1 ATCACTGTGCTCCGAGGCAGCCCCGATAGCTCCTCCAACTCCAGCCACAGCCTCATACAG ism1_T3_emb1.1 ATCACTGTGCTCCGAG------------------------CCAGCCACAGCCTCATACAG ism1_T3_emb1.2 ATCACTGTGCTCCGAGGCAGCCCCGAT----------ACTCCAGCCACAGCCTCATACAG ism1_T3_emb1.3 GTCAGTCCATTCCATTCCA---CATACAGGTCAGTCCATTCCACATACAGGT-----CAG ism1_T3_emb1.4 GTCAGTCCATTCCATTCCA---CATACAGGTCAGTCCATTCCACATACAGGT-----CAG ism1 943 GTCAGTCCATTCCACATACAG 964 uninjected_emb1 GTCAGTCCATTCCTCATACAG ism1_T3_emb1.1 GTCAGTCCATTCCACATACAG ism1_T3_emb1.2 GTCAGTCCATTCCACATACAG ism1_T3_emb1.3 TCCATTCCATTCCACATACAG ism1_T3_emb1.4 TCCATTCCATTCCACATACAG mRNA ism1 1 MLRLAAELLLLLGLLLLTLHITVLRG^SPDSSSNSSHSLIQVSPFHIQ… uninjected_emb1 MLRLAAELLLLLGLLLLTLHITVLRG^SPDSSSNSSHSLIQVSPFHIQ… ism1_T3_emb1.1 MLRLAAELLLLLGLLLLTLHITVLRV-------SSHSLIQVSPFHIQ… ism1_T3_emb1.2 MLRLAAELLLLLGLLLLTLHITVLRG^SPD---YSSHSLIQVSPFHIQ… ism1_T3_emb1.3 MLRLAAELLLLLGLLLLLIQVSP^FHSISYRSVHSTYRF-QSIPFHIQ ism1_T3_emb1.4 MLRLAAELLLLLGLLLLLIQVSP^FHSISYRSVHSTYRF-QSIPFHIQ Human ISM1 MVRLAAELLLLLGLLLLTLHITVLRG^SGA -

Role of Genomic Variants in the Response to Biologics Targeting Common Autoimmune Disorders
Role of genomic variants in the response to biologics targeting common autoimmune disorders by Gordana Lenert, PhD The thesis submitted to the Faculty of Graduate and Postdoctoral Affairs in partial fulfillment of the requirements for the degree of Master of Science Ottawa-Carleton Joint Program in Bioinformatics Carleton University Ottawa, Canada © 2016 Gordana Lenert Abstract Autoimmune diseases (AID) are common chronic inflammatory conditions initiated by the loss of the immunological tolerance to self-antigens. Chronic immune response and uncontrolled inflammation provoke diverse clinical manifestations, causing impairment of various tissues, organs or organ systems. To avoid disability and death, AID must be managed in clinical practice over long periods with complex and closely controlled medication regimens. The anti-tumor necrosis factor biologics (aTNFs) are targeted therapeutic drugs used for AID management. However, in spite of being very successful therapeutics, aTNFs are not able to induce remission in one third of AID phenotypes. In our research, we investigated genomic variability of AID phenotypes in order to explain unpredictable lack of response to aTNFs. Our hypothesis is that key genetic factors, responsible for the aTNFs unresponsiveness, are positioned at the crossroads between aTNF therapeutic processes that generate remission and pathogenic or disease processes that lead to AID phenotypes expression. In order to find these key genetic factors at the intersection of the curative and the disease pathways, we combined genomic variation data collected from publicly available curated AID genome wide association studies (AID GWAS) for each disease. Using collected data, we performed prioritization of genes and other genomic structures, defined the key disease pathways and networks, and related the results with the known data by the bioinformatics approaches. -
Figure S1. Reverse Transcription‑Quantitative PCR Analysis of ETV5 Mrna Expression Levels in Parental and ETV5 Stable Transfectants
Figure S1. Reverse transcription‑quantitative PCR analysis of ETV5 mRNA expression levels in parental and ETV5 stable transfectants. (A) Hec1a and Hec1a‑ETV5 EC cell lines; (B) Ishikawa and Ishikawa‑ETV5 EC cell lines. **P<0.005, unpaired Student's t‑test. EC, endometrial cancer; ETV5, ETS variant transcription factor 5. Figure S2. Survival analysis of sample clusters 1‑4. Kaplan Meier graphs for (A) recurrence‑free and (B) overall survival. Survival curves were constructed using the Kaplan‑Meier method, and differences between sample cluster curves were analyzed by log‑rank test. Figure S3. ROC analysis of hub genes. For each gene, ROC curve (left) and mRNA expression levels (right) in control (n=35) and tumor (n=545) samples from The Cancer Genome Atlas Uterine Corpus Endometrioid Cancer cohort are shown. mRNA levels are expressed as Log2(x+1), where ‘x’ is the RSEM normalized expression value. ROC, receiver operating characteristic. Table SI. Clinicopathological characteristics of the GSE17025 dataset. Characteristic n % Atrophic endometrium 12 (postmenopausal) (Control group) Tumor stage I 91 100 Histology Endometrioid adenocarcinoma 79 86.81 Papillary serous 12 13.19 Histological grade Grade 1 30 32.97 Grade 2 36 39.56 Grade 3 25 27.47 Myometrial invasiona Superficial (<50%) 67 74.44 Deep (>50%) 23 25.56 aMyometrial invasion information was available for 90 of 91 tumor samples. Table SII. Clinicopathological characteristics of The Cancer Genome Atlas Uterine Corpus Endometrioid Cancer dataset. Characteristic n % Solid tissue normal 16 Tumor samples Stagea I 226 68.278 II 19 5.740 III 70 21.148 IV 16 4.834 Histology Endometrioid 271 81.381 Mixed 10 3.003 Serous 52 15.616 Histological grade Grade 1 78 23.423 Grade 2 91 27.327 Grade 3 164 49.249 Molecular subtypeb POLE 17 7.328 MSI 65 28.017 CN Low 90 38.793 CN High 60 25.862 CN, copy number; MSI, microsatellite instability; POLE, DNA polymerase ε. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Downloaded from Ensembl
UCSF UC San Francisco Electronic Theses and Dissertations Title Detecting genetic similarity between complex human traits by exploring their common molecular mechanism Permalink https://escholarship.org/uc/item/1k40s443 Author Gu, Jialiang Publication Date 2019 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California by Submitted in partial satisfaction of the requirements for degree of in in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO AND UNIVERSITY OF CALIFORNIA, BERKELEY Approved: ______________________________________________________________________________ Chair ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ Committee Members ii Acknowledgement This project would not have been possible without Prof. Dr. Hao Li, Dr. Jiashun Zheng and Dr. Chris Fuller at the University of California, San Francisco (UCSF) and Caribou Bioscience. The Li lab grew into a multi-facet research group consist of both experimentalists and computational biologists covering three research areas including cellular/molecular mechanism of ageing, genetic determinants of complex human traits and structure, function, evolution of gene regulatory network. Labs like these are the pillar of global success and reputation -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Table 3 Two-Class Paired Significance Analysis of Microarrays Comparing Gene Expression Between Paired
Supplemental Table 3 Two‐class paired Significance Analysis of Microarrays comparing gene expression between paired pre‐ and post‐transplant kidneys biopsies (N=8). Entrez Fold q‐value Probe Set ID Gene Symbol Unigene Name Score Gene ID Difference (%) Probe sets higher expressed in post‐transplant biopsies in paired analysis (N=1871) 218870_at 55843 ARHGAP15 Rho GTPase activating protein 15 7,01 3,99 0,00 205304_s_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 6,30 4,50 0,00 1563649_at ‐‐ ‐‐ ‐‐ 6,24 3,51 0,00 1567913_at 541466 CT45‐1 cancer/testis antigen CT45‐1 5,90 4,21 0,00 203932_at 3109 HLA‐DMB major histocompatibility complex, class II, DM beta 5,83 3,20 0,00 204606_at 6366 CCL21 chemokine (C‐C motif) ligand 21 5,82 10,42 0,00 205898_at 1524 CX3CR1 chemokine (C‐X3‐C motif) receptor 1 5,74 8,50 0,00 205303_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 5,68 6,87 0,00 226841_at 219972 MPEG1 macrophage expressed gene 1 5,59 3,76 0,00 203923_s_at 1536 CYBB cytochrome b‐245, beta polypeptide (chronic granulomatous disease) 5,58 4,70 0,00 210135_s_at 6474 SHOX2 short stature homeobox 2 5,53 5,58 0,00 1562642_at ‐‐ ‐‐ ‐‐ 5,42 5,03 0,00 242605_at 1634 DCN decorin 5,23 3,92 0,00 228750_at ‐‐ ‐‐ ‐‐ 5,21 7,22 0,00 collagen, type III, alpha 1 (Ehlers‐Danlos syndrome type IV, autosomal 201852_x_at 1281 COL3A1 dominant) 5,10 8,46 0,00 3493///3 IGHA1///IGHA immunoglobulin heavy constant alpha 1///immunoglobulin heavy 217022_s_at 494 2 constant alpha 2 (A2m marker) 5,07 9,53 0,00 1 202311_s_at -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -
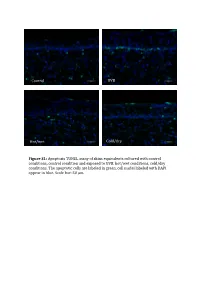
Supplemental Data and Tables
Control UVB Hot/wet Cold/dry Figure S1: Apoptosis TUNEL assay of skins equivalents cultured with control conditions, control condition and exposed to UVB, hot/wet conditions, cold/dry conditions. The apoptotic cells are laBeled in green, cell nuclei laBeled with DAPI appear in blue. Scale bar: 50 μm. Table SI: list of oligonucleotides used in QPCR experiments Gene Forward primer Reverse primer LCE1A TGCAAGAGTGGCTGAGATGC AGACAACACAGTTGGTGT LCE1DE TGAATAGCTGAGAGGTTC CAGCCATGGATCTGCAGAAG LCE5A CTGATGCTAGGTCAGGACTGA TGGTCCTGAGAAGCACTCTAC COL4A1 CCAGGATTTCAAGGTCCAAA CTCCCCTTTGATGATGTCGT LUM TGGCATTGATTGGTGGTACCA TGGGTAGCTTTCAGGGCAGTT FOS CAAGCGGAGACAGACCAACT AGTCAGATCAAGGGAAGCCA Table SII – Part I: genes significantly modulated after treatment 1 (hot-wet) p-value Fold change Gene Symbol Description (Treat-1_vs_CTRL) (Treat-1_vs_CTRL) 0,001147884 15,61495918 MIR639 Homo sapiens microRNA 639 (MIR639), microRNA. 0,021880609 12,6104707 LOC642685 PREDICTED: Homo sapiens similar to maternal G10 transcript (LOC642685), mRNA. 0,010879477 8,858661664 LOC389365 PREDICTED: Homo sapiens hypothetical LOC389365 (LOC389365), mRNA. 0,022200861 6,339924039 HS.566764 DKFZp781A1072_s1 781 (synonym: hlcc4) Homo sapiens cDNA clone DKFZp781A1072 3, mRNA sequence 0,022672172 5,826114515 DKK4 Homo sapiens dickkopf homolog 4 (Xenopus laevis) (DKK4), mRNA. 0,036661342 5,448113208 SLC25A29 Homo sapiens solute carrier family 25, member 29 (SLC25A29), nuclear gene encoding mitochondrial protein, mRNA. 0,035172618 5,141273735 LOC100128361 PREDICTED: Homo sapiens similar to hCG1985411 (LOC100128361), mRNA. 0,01471108 4,783488094 LOC652119 PREDICTED: Homo sapiens similar to putative DUX4 protein (LOC652119), mRNA. 0,009233667 4,511367872 REPS1 Homo sapiens RALBP1 associated Eps domain containing 1 (REPS1), mRNA. 0,031513664 4,407256363 LOC283314 PREDICTED: Homo sapiens misc_RNA (LOC283314), miscRNA. 0,010390588 3,970881226 LOC642773 PREDICTED: Homo sapiens similar to Myc-associated zinc finger protein (MAZI) 0,006806623 3,764373992 LOC391132 PREDICTED: Homo sapiens misc_RNA (LOC391132), miscRNA. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 27, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 27, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN.