Selectivity Optimization in Tandem Column Liquid Chromatography
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

Prohibited Substances List
Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). Neither the List nor the EADCM Regulations are in current usage. Both come into effect on 1 January 2010. The current list of FEI prohibited substances remains in effect until 31 December 2009 and can be found at Annex II Vet Regs (11th edition) Changes in this List : Shaded row means that either removed or allowed at certain limits only SUBSTANCE ACTIVITY Banned Substances 1 Acebutolol Beta blocker 2 Acefylline Bronchodilator 3 Acemetacin NSAID 4 Acenocoumarol Anticoagulant 5 Acetanilid Analgesic/anti-pyretic 6 Acetohexamide Pancreatic stimulant 7 Acetominophen (Paracetamol) Analgesic/anti-pyretic 8 Acetophenazine Antipsychotic 9 Acetylmorphine Narcotic 10 Adinazolam Anxiolytic 11 Adiphenine Anti-spasmodic 12 Adrafinil Stimulant 13 Adrenaline Stimulant 14 Adrenochrome Haemostatic 15 Alclofenac NSAID 16 Alcuronium Muscle relaxant 17 Aldosterone Hormone 18 Alfentanil Narcotic 19 Allopurinol Xanthine oxidase inhibitor (anti-hyperuricaemia) 20 Almotriptan 5 HT agonist (anti-migraine) 21 Alphadolone acetate Neurosteriod 22 Alphaprodine Opiod analgesic 23 Alpidem Anxiolytic 24 Alprazolam Anxiolytic 25 Alprenolol Beta blocker 26 Althesin IV anaesthetic 27 Althiazide Diuretic 28 Altrenogest (in males and gelidngs) Oestrus suppression 29 Alverine Antispasmodic 30 Amantadine Dopaminergic 31 Ambenonium Cholinesterase inhibition 32 Ambucetamide Antispasmodic 33 Amethocaine Local anaesthetic 34 Amfepramone Stimulant 35 Amfetaminil Stimulant 36 Amidephrine Vasoconstrictor 37 Amiloride Diuretic 1 Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). -
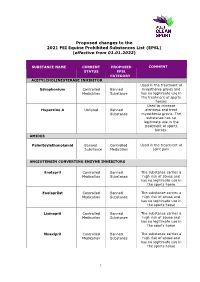
Proposed Changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (Effective from 01.01.2022)
Proposed changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (effective from 01.01.2022) SUBSTANCE NAME CURRENT PROPOSED COMMENT STATUS EPSL CATEGORY ACETYLCHOLINESTERASE INHIBITOR Used in the treatment of Edrophonium Controlled Banned myasthenia gravis and Medication Substance has no legitimate use in the treatment of sports horses Used to increase Huperzine A Unlisted Banned alertness and treat Substance myasthenia gravis. The substance has no legitimate use in the treatment of sports horses. AMIDES Palmitoylethanolamid Banned Controlled Used in the treatment of Substance Medication joint pain ANGIOTENSIN CONVERTING ENZYME INHIBITORS Enalapril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Enalaprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Lisinopril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Moexipril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse 1 Perindoprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse ANTIHISTAMINES Antazoline Controlled Banned The substance has no Medication Substance legitimate use in the sports horse Azatadine Controlled Banned The substance has Medication Substance sedative effects -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] BMJ Open Pediatric drug utilization in the Western Pacific region: Australia, Japan, South Korea, Hong Kong and Taiwan Journal: BMJ Open ManuscriptFor ID peerbmjopen-2019-032426 review only Article Type: Research Date Submitted by the 27-Jun-2019 Author: Complete List of Authors: Brauer, Ruth; University College London, Research Department of Practice and Policy, School of Pharmacy Wong, Ian; University College London, Research Department of Practice and Policy, School of Pharmacy; University of Hong Kong, Centre for Safe Medication Practice and Research, Department -

(12) Patent Application Publication (10) Pub. No.: US 2002/0102215 A1 100 Ol
US 2002O102215A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2002/0102215 A1 Klaveness et al. (43) Pub. Date: Aug. 1, 2002 (54) DIAGNOSTIC/THERAPEUTICAGENTS (60) Provisional application No. 60/049.264, filed on Jun. 6, 1997. Provisional application No. 60/049,265, filed (75) Inventors: Jo Klaveness, Oslo (NO); Pal on Jun. 6, 1997. Provisional application No. 60/049, Rongved, Oslo (NO); Anders Hogset, 268, filed on Jun. 7, 1997. Oslo (NO); Helge Tolleshaug, Oslo (NO); Anne Naevestad, Oslo (NO); (30) Foreign Application Priority Data Halldis Hellebust, Oslo (NO); Lars Hoff, Oslo (NO); Alan Cuthbertson, Oct. 28, 1996 (GB)......................................... 9622.366.4 Oslo (NO); Dagfinn Lovhaug, Oslo Oct. 28, 1996 (GB). ... 96223672 (NO); Magne Solbakken, Oslo (NO) Oct. 28, 1996 (GB). 9622368.0 Jan. 15, 1997 (GB). ... 97OO699.3 Correspondence Address: Apr. 24, 1997 (GB). ... 9708265.5 BACON & THOMAS, PLLC Jun. 6, 1997 (GB). ... 9711842.6 4th Floor Jun. 6, 1997 (GB)......................................... 97.11846.7 625 Slaters Lane Alexandria, VA 22314-1176 (US) Publication Classification (73) Assignee: NYCOMED IMAGING AS (51) Int. Cl." .......................... A61K 49/00; A61K 48/00 (52) U.S. Cl. ............................................. 424/9.52; 514/44 (21) Appl. No.: 09/765,614 (22) Filed: Jan. 22, 2001 (57) ABSTRACT Related U.S. Application Data Targetable diagnostic and/or therapeutically active agents, (63) Continuation of application No. 08/960,054, filed on e.g. ultrasound contrast agents, having reporters comprising Oct. 29, 1997, now patented, which is a continuation gas-filled microbubbles stabilized by monolayers of film in-part of application No. 08/958,993, filed on Oct. -

2 12/ 35 74Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 22 March 2012 (22.03.2012) 2 12/ 35 74 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 9/16 (2006.01) A61K 9/51 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 9/14 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) International Application Number: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/EP201 1/065959 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 14 September 201 1 (14.09.201 1) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, (25) Filing Language: English RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, (26) Publication Language: English TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/382,653 14 September 2010 (14.09.2010) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, NANOLOGICA AB [SE/SE]; P.O Box 8182, S-104 20 ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, Stockholm (SE). -

Final1-Aritmo-Final-Report-V2-0Final.Pdf
ARITMO Final Report PROJECT FINAL REPORT Grant Agreement number: 241679 Project acronym: ARITMO Project title: Arrhythmogenic potential of drugs Funding Scheme: Small or Medium-Scale Focused Research Project Period covered: from 1st January 2010 to 30th June 2013 Name of the scientific representative of the project's co-ordinator, Title and Organisation: Prof. Miriam CJM Sturkenboom, Erasmus Universitair Medisch Centrum Rotterdam Tel: +31 10 704 4126 Fax: +31 10 704 4722 E-mail: [email protected] Project website1 address: www.aritmo-project.org 1 The home page of the website should contain the generic European flag and the FP7 logo which are available in electronic format at the Europa website (logo of the European flag: http://europa.eu/abc/symbols/emblem/index_en.htm ; logo of the 7th FP: http://ec.europa.eu/research/fp7/index_en.cfm?pg=logos). The area of activity of the project should also be mentioned. © Copyright 2013 ARITMO Consortium 1 ARITMO Final Report Table of contents Table of contents ................................................................................................................................................................. 2 1. Final publishable summary report ................................................................................................................................ 3 1.1 Executive summary ................................................................................................................................................. 3 1.2 Description of project context and -

Common Study Protocol for Observational Database Studies WP5 – Analytic Database Studies
Arrhythmogenic potential of drugs FP7-HEALTH-241679 http://www.aritmo-project.org/ Common Study Protocol for Observational Database Studies WP5 – Analytic Database Studies V 1.3 Draft Lead beneficiary: EMC Date: 03/01/2010 Nature: Report Dissemination level: D5.2 Report on Common Study Protocol for Observational Database Studies WP5: Conduct of Additional Observational Security: Studies. Author(s): Gianluca Trifiro’ (EMC), Giampiero Version: v1.1– 2/85 Mazzaglia (F-SIMG) Draft TABLE OF CONTENTS DOCUMENT INFOOMATION AND HISTORY ...........................................................................4 DEFINITIONS .................................................... ERRORE. IL SEGNALIBRO NON È DEFINITO. ABBREVIATIONS ......................................................................................................................6 1. BACKGROUND .................................................................................................................7 2. STUDY OBJECTIVES................................ ERRORE. IL SEGNALIBRO NON È DEFINITO. 3. METHODS ..........................................................................................................................8 3.1.STUDY DESIGN ....................................................................................................................8 3.2.DATA SOURCES ..................................................................................................................9 3.2.1. IPCI Database .....................................................................................................9 -

Anticholinergic Syndrome 23 William J
Anticholinergic Syndrome 23 William J. Boroughf Contents The anticholinergic syndrome is common and Overview and Incidence ............................ 521 may result from exposures to many drugs or nat- ural substances (Table 1). Anticholinergic effects History ............................................... 521 are desired or intended effects for certain drugs Pathophysiology .................................... 522 (i.e., antispasmodics, mydriatics, and belladonna Toxic Mechanism .................................... 523 alkaloids) and are undesired or side-effects for Clinical Presentations and Life-Threatening other drugs (i.e., antihistamines, antidepressants, Complications ....................................... 525 antipsychotics, and antiparkinsonians). Both pre- Routes of Exposure .................................. 526 Intention or Cause of Exposures ..................... 526 scription and over-the-counter drugs may have Range of Toxicity .................................... 527 anticholinergic effects. Combined use of more Organ System Effects ................................ 527 than one drug with anticholinergic effects Diagnosis ............................................ 529 increases the risk of anticholinergic toxicity. The Laboratory Tests ..................................... 529 anticholinergic syndrome, also called the anticho- Differential Diagnosis . ............................. 530 linergic toxidrome, has peripheral and central Diagnostic Studies ................................... 531 manifestations. The more -

2021 Equine Prohibited Substances List
2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. LISTED AS SUBSTANCE ACTIVITY BANNED 1-androsterone Anabolic BANNED 3β-Hydroxy-5α-androstan-17-one Anabolic BANNED 4-chlorometatandienone Anabolic BANNED 5α-Androst-2-ene-17one Anabolic BANNED 5α-Androstane-3α, 17α-diol Anabolic BANNED 5α-Androstane-3α, 17β-diol Anabolic BANNED 5α-Androstane-3β, 17α-diol Anabolic BANNED 5α-Androstane-3β, 17β-diol Anabolic BANNED 5β-Androstane-3α, 17β-diol Anabolic BANNED 7α-Hydroxy-DHEA Anabolic BANNED 7β-Hydroxy-DHEA Anabolic BANNED 7-Keto-DHEA Anabolic CONTROLLED 17-Alpha-Hydroxy Progesterone Hormone FEMALES BANNED 17-Alpha-Hydroxy Progesterone Anabolic MALES BANNED 19-Norandrosterone Anabolic BANNED 19-Noretiocholanolone Anabolic BANNED 20-Hydroxyecdysone Anabolic BANNED Δ1-Testosterone Anabolic BANNED Acebutolol Beta blocker BANNED Acefylline Bronchodilator BANNED Acemetacin Non-steroidal anti-inflammatory drug BANNED Acenocoumarol Anticoagulant CONTROLLED Acepromazine Sedative BANNED Acetanilid Analgesic/antipyretic CONTROLLED Acetazolamide Carbonic Anhydrase Inhibitor BANNED Acetohexamide Pancreatic stimulant CONTROLLED Acetominophen (Paracetamol) Analgesic BANNED Acetophenazine Antipsychotic BANNED Acetophenetidin (Phenacetin) Analgesic BANNED Acetylmorphine Narcotic BANNED Adinazolam Anxiolytic BANNED Adiphenine Antispasmodic BANNED Adrafinil Stimulant 1 December 2020, Lausanne, Switzerland 2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). -

Elective Testing Substances List
ELECTIVE TESTING SUBSTANCES The FEI Approved Laboratories will undertake Elective Testing for up to four of the following substances. SUBSTANCE ACTIVITY 17-Alpha-Hydroxy Progesterone FEMALES Hormone Acepromazine Sedative Acetazolamide Carbonic Anhydrase Inhibitor Acetominophen (Paracetamol) Analgesic Adrenaline Stimulant Adrenocorticotropic hormone (ACTH) Hormone Aformoterol Bronchodilator Albuterol (Salbutamol) Bronchodilator Alpha-Casozepine Sedative Altrenogest (in males and geldings) Oestrus suppression Amantadine Dopaminergic Ambroxol Mucolytic Amcinonide Corticosteroid Aminocaproic acid Haemostatic (anti-fibrinolytic) Aminophylline Bronchodilator Aminorex Parasympathomimetic Amiodarone Anti-arrhythmic Antazoline Antihistamine Atipamezole Alpha adrenergic antagonist Atropine (Specified Substance) Anticholinergic Azatadine Antihistamine Beclomethasone Corticosteroid Benzocaine (Ethyl Aminobenzoate) Local anaesthetic Benzquinamide Antihistamine Benzydamine Non-steroidal anti-inflammatory drug Betamethasone Corticosteroid Last Updated: 1 December 2020 Bethanechol Parasympathetic agonist Brinzolamide Diuretic Bromhexine Mucolytic Bromodiphenhydramine Antihistamine Brompheniramine Antihistamine Buclizine Antihistamine Budesonide Corticosteroid Bupivacaine Local anaesthetic Buprenorphine Opioid Butorphanol Opioid Butylscopolamine Anticholinergic Caffeine (Specified Substance) Central nervous system stimulant Calcium dobesilate Vasotropic Captopril Angiotensin-converting enzyme inhibitor Carbamazepine Anticonvulsant Carbon dioxide (CO2) -

Ep 2301628 A1
(19) & (11) EP 2 301 628 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 30.03.2011 Bulletin 2011/13 A61P 37/06 (2006.01) (21) Application number: 10013062.4 (22) Date of filing: 13.10.2004 (84) Designated Contracting States: • Manivasakam, Palaniyandi AT BE BG CH CY CZ DE DK EE ES FI FR GB GR West Roxbury HU IE IT LI LU MC NL PL PT RO SE SI SK TR MA 02132 (US) Designated Extension States: • Hurst, Nicole AL HR LT LV MK Boston MA 02132 (US) (30) Priority: 15.10.2003 US 512415 P • Foley, Michael A. Chestnut Hill (62) Document number(s) of the earlier application(s) in MA (US) accordance with Art. 76 EPC: • Slavonic, Michael S. 09002049.6 / 2 070 550 Quincy 04809944.4 / 1 680 121 MA 02169 (US) • Smith, Brendan (71) Applicant: Zalicus Inc. Sommerville Cambridge, MA 02142 (US) MA 02118 (US) • Auspitz, Benjamin A. (72) Inventors: Cambridge • Keith, Curtis MA 02139 (US) Boston MA 02118 (US) (74) Representative: Bösl, Raphael Konrad • Borisy, Alexis Isenbruck Bösl Hörschler LLP Arlington Patentanwälte MA 02476 (US) Prinzregentenstrasse 68 • Zimmermann, Grant R. 81675 München (DE) Winchester MA 01890 (US) Remarks: •Jost-Price, Edward Roydon This application was filed on 01-20-2010 as a West Roxbury divisional application to the application mentioned MA 02132 (US) under INID code 62. (54) Methods and reagents for the treatment of immunoinflammatory disorders (57) The invention features, among others, a tetra- stituted pyrimidopyrimidine and a corticosteroid for use substituted pyrimidopyrimidine and a corticosteroid for ina method for treating an immunoinflammatory disorder.