Comparative DNA Methylation Profiling Reveals An
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In Silico Analysis Identifies Genes Common Between Five Primary Gastrointestinal Cancer Sites with Potential Clinical Applications
ORIGINAL ARTICLE Annals of Gastroenterology (2014) 27, 1-14 In silico analysis identifies genes common between five primary gastrointestinal cancer sites with potential clinical applications Subhankar Chakraborty University of Nebraska Medical Center, Omaha, NE, USA Abstract Background Previous studies have investigated differential gene expression in gastrointes- tinal (GI) epithelial cancers by microarray. The aim of the present study was to use data from the Oncomine database to identify genes that share a similar differential expression in two or more primary GI cancer sites. Methods Five thousand of the most differentially expressed genes in epithelial cancers (com- pared to normal tissue) arising in the pancreas, liver, stomach, esophagus or colorectum were identified (1,000 per primary site) from Oncomine. Using Venn diagrams, genes common to two or more primary GI sites were identified. Functional and pathway analysis was performed on genes that were similarly expressed in ≥3 of the five areas of the GI tract. Results Forty six studies comprising 5,876 samples were included. Overall, 90.6% genes were unique to the respective primary sites, 7.4% shared between two GI primary sites, 1.8% between three and 0.2% between four GI primary sites. Pancreatic and hepatocellular cancers (HCC) shared most number of upregulated genes (N=66) while HCC and gastric cancer shared most downregulated genes (N=59). Genes encoding enzymes comprised the most commonly shared genes between GI primary sites (30.4% of upregulated and 63.2% of downregulated genes). Those genes that were shared between three or more GI primary sites also showed significant differential expression in the same direction in other non-GI cancers. -

Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis
Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma, Anna-Karin Ekman, Cecilia Bivik Eding and Charlotta Enerbäck The self-archived postprint version of this journal article is available at Linköping University Institutional Repository (DiVA): http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-147791 N.B.: When citing this work, cite the original publication. Verma, D., Ekman, A., Bivik Eding, C., Enerbäck, C., (2018), Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis, Journal of Investigative Dermatology, 138(5), 1088-1093. https://doi.org/10.1016/j.jid.2017.11.036 Original publication available at: https://doi.org/10.1016/j.jid.2017.11.036 Copyright: Elsevier http://www.elsevier.com/ Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma*a, Anna-Karin Ekman*a, Cecilia Bivik Edinga and Charlotta Enerbäcka *Authors contributed equally aIngrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Division of Dermatology, Linköping University, Linköping, Sweden Corresponding author: Charlotta Enerbäck Ingrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Linköping University SE-581 85 Linköping, Sweden Phone: +46 10 103 7429 E-mail: [email protected] Short title Differential methylation in psoriasis Abbreviations CGI, CpG island; DMS, differentially methylated site; RRBS, reduced representation bisulphite sequencing Keywords (max 6) psoriasis, epidermis, methylation, Wnt, susceptibility, expression 1 ABSTRACT Psoriasis is a chronic inflammatory skin disease with both local and systemic components. Genome-wide approaches have identified more than 60 psoriasis-susceptibility loci, but genes are estimated to explain only one third of the heritability in psoriasis, suggesting additional, yet unidentified, sources of heritability. -
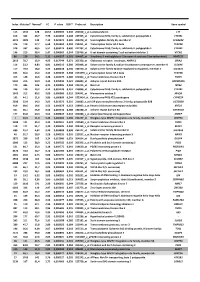
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244 -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Open Sadiesteffens Dissertation Final.Pdf
The Pennsylvania State University The Graduate School College of Medicine MECHANISM OF DRUG ACTION OF THE SPECIFIC CK2 INHIBITOR CX-4945 IN ACUTE MYELOID LEUKEMIA A Dissertation in Biomedical Sciences by Sadie Lynne Steffens 2015 Sadie Lynne Steffens Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2015 The dissertation of Sadie Lynne Steffens was reviewed and approved* by the following: Sinisa Dovat Physician, Associate Professor of Pediatrics, Pharmacology, & Biochemistry Director, Translational Research – Four Diamonds Pediatric Cancer Research Center Dissertation Advisor Chair of Committee Barbara A. Miller Physician, Professor of Pediatrics Chief, Division of Pediatric Hematology/Oncology Sergei A. Grigoryev Professor of Biochemistry and Molecular Biology Jong K. Yun Associate Professor of Pharmacology Ralph L. Keil Associate Professor of Biochemistry and Molecular Biology Chair, Biomedical Sciences Graduate Program *Signatures are on file in the Graduate School ii ABSTRACT Acute myeloid leukemia (AML) is a malignant disease of the myeloid line of blood cells and is characterized by the rapid growth of abnormal white blood cells that accumulate in the bone marrow and interfere with the production of normal blood cells. Cytarabine and other currently available treatments for acute myeloid leukemia are highly toxic and insufficient, as more than half of all AML patients develop resistance to chemotherapeutic agents. Since AML often affects older people who are less tolerant of chemotherapy, there is need for novel, targeted, less toxic drugs in order to improve survival for this disease. Casein Kinase II (CK2) is a pro-oncogenic serine/threonine kinase that is essential for cellular proliferation. -

Product Datasheet KIAA1704 Antibody NBP1
Product Datasheet KIAA1704 Antibody NBP1-79666 Unit Size: 100 ul Store at -20C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBP1-79666 Updated 12/9/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBP1-79666 Page 1 of 2 v.20.1 Updated 12/9/2020 NBP1-79666 KIAA1704 Antibody Product Information Unit Size 100 ul Concentration Please see the vial label for concentration. If unlisted please contact technical services. Storage Store at -20C. Avoid freeze-thaw cycles. Clonality Polyclonal Preservative 0.09% Sodium Azide Isotype IgG Purity Immunogen affinity purified Buffer PBS and 2% Sucrose Product Description Host Rabbit Gene ID 55425 Gene Symbol GPALPP1 Species Human Immunogen Synthetic peptide directed towards the middle region of human KIAA1704The immunogen for this antibody is KIAA1704. Peptide sequence KAAEDKNKPQERIPFDRDKDLKVNRFDEAQKKALIKKSRELNTRFSHGKG. The peptide sequence for this immunogen was taken from within the described region. Notes The addition of 50% glycerol is optional for those storing this antibody at -20C and not aliquoting smaller units. However, please note that glycerol may interrupt some downstream antibody applications and should be added with caution. Product Application Details Applications Western Blot Recommended Dilutions Western Blot 1:1000 Application Notes This is a rabbit polyclonal antibody against KIAA1704 and was validated on Western blot. Images Western Blot: KIAA1704 Antibody [NBP1-79666] - Human Spleen lysate, concentration 0.2-1 ug/ml. Novus Biologicals USA Bio-Techne Canada 10730 E. -

Genome-Wide CRISPR-Cas9 Screens Reveal Loss of Redundancy Between PKMYT1 and WEE1 in Glioblastoma Stem-Like Cells
Article Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells Graphical Abstract Authors Chad M. Toledo, Yu Ding, Pia Hoellerbauer, ..., Bruce E. Clurman, James M. Olson, Patrick J. Paddison Correspondence [email protected] (J.M.O.), [email protected] (P.J.P.) In Brief Patient-derived glioblastoma stem-like cells (GSCs) can be grown in conditions that preserve patient tumor signatures and their tumor initiating capacity. Toledo et al. use these conditions to perform genome-wide CRISPR-Cas9 lethality screens in both GSCs and non- transformed NSCs, revealing PKMYT1 as a candidate GSC-lethal gene. Highlights d CRISPR-Cas9 lethality screens performed in patient brain- tumor stem-like cells d PKMYT1 is identified in GSCs, but not NSCs, as essential for facilitating mitosis d PKMYT1 and WEE1 act redundantly in NSCs, where their inhibition is synthetic lethal d PKMYT1 and WEE1 redundancy can be broken by over- activation of EGFR and AKT Toledo et al., 2015, Cell Reports 13, 2425–2439 December 22, 2015 ª2015 The Authors http://dx.doi.org/10.1016/j.celrep.2015.11.021 Cell Reports Article Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells Chad M. Toledo,1,2,14 Yu Ding,1,14 Pia Hoellerbauer,1,2 Ryan J. Davis,1,2,3 Ryan Basom,4 Emily J. Girard,3 Eunjee Lee,5 Philip Corrin,1 Traver Hart,6,7 Hamid Bolouri,1 Jerry Davison,4 Qing Zhang,4 Justin Hardcastle,1 Bruce J. Aronow,8 Christopher L. -

Reduced-Representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-Scale Comparison of Drug-Induced Phenotypes*□S
crossmark Research © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes*□S Jennifer G. Abelin‡ ‡‡, Jinal Patel‡ ‡‡, Xiaodong Lu‡ ‡‡, Caitlin M. Feeney‡ ‡‡, Lola Fagbami‡, Amanda L. Creech‡, Roger Hu‡, Daniel Lam‡, Desiree Davison‡, Lindsay Pino‡, Jana W. Qiao‡, Eric Kuhn‡, Adam Officer‡, Jianxue Li§, Susan Abbatiello‡, Aravind Subramanian‡, Richard Sidman§, Evan Snyder¶, Steven A. Carr‡, and Jacob D. Jaffe‡ʈ Profiling post-translational modifications represents an duced-representation phosphopeptide probes to be de- alternative dimension to gene expression data in charac- tected in ϳ200 samples. terizing cellular processes. Many cellular responses to The performance of the assay was evaluated by mea- drugs are mediated by changes in cellular phosphosignal- suring the probes in new samples generated under treat- ing. We sought to develop a common platform on which ment conditions from discovery experiments, recapitulating phosphosignaling responses could be profiled across the observations of deeper experiments using a fraction thousands of samples, and created a targeted MS assay of the analytical effort. We measured these probes in new that profiles a reduced-representation set of phospho- experiments varying the treatments, cell types, and time- peptides that we show to be strong indicators of re- points to demonstrate generalizability. We demonstrated sponses to chemical perturbagens. that the assay is sensitive to disruptions in common sig- To develop the assay, we investigated the coordinate naling pathways (e.g. MAPK, PI3K/mTOR, and CDK). -

The Genetics of Heterotaxy Syndrome
The Genetics of Heterotaxy Syndrome A dissertation submitted to the Division of Graduate Studies and Research, University of Cincinnati in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Molecular and Developmental Biology by Jason R. Cowan Master of Science, University of British Columbia, Vancouver, 2007 Committee Chair: Stephanie M. Ware, M.D., Ph.D. Robert B. Hinton Jr., M.D. Linda M. Parysek, Ph.D. S. Steven Potter, Ph.D. Aaron M. Zorn, Ph.D. Molecular & Developmental Biology Graduate Program College of Medicine, University of Cincinnati Division of Developmental Biology, Cincinnati Children’s Hospital Medical Center Cincinnati, Ohio, USA, 2015 ABSTRACT Congenital heart defects (CHDs) are the greatest cause of infant morbidity and mortality worldwide, occurring in roughly 8 per 1000 live births (~1%). Heterotaxy, a multiple congenital anomaly syndrome resulting from failure to establish left-right (L-R) asymmetry, is characterized by diverse, complex CHDs. Heterogeneous in presentation and etiology, heterotaxy serves as a complex and growing focal point for cardiovascular genetic research. In the two decades since the zinc finger transcription factor, ZIC3, was first identified as a cause of X-linked heterotaxy, mutations in nearly twenty genes with L-R patterning functions have been detected among patients with heterotaxy. Nevertheless, despite considerable progress, genetic causes for heterotaxy remain largely uncharacterized. With an estimated 70-80% of heterotaxy cases still unexplained, there remains enormous potential for novel gene discovery. In this dissertation, we have balanced gene discovery efforts aimed at identifying and characterizing novel causes of heterotaxy with studies into the mechanisms governing ZIC3- related heterotaxy. -

Lupus Nephritis Supp Table 5
Supplementary Table 5 : Transcripts and DAVID pathways correlating with the expression of CD4 in lupus kidney biopsies Positive correlation Negative correlation Transcripts Pathways Transcripts Pathways Identifier Gene Symbol Correlation coefficient with CD4 Annotation Cluster 1 Enrichment Score: 26.47 Count P_Value Benjamini Identifier Gene Symbol Correlation coefficient with CD4 Annotation Cluster 1 Enrichment Score: 3.16 Count P_Value Benjamini ILMN_1727284 CD4 1 GOTERM_BP_FAT translational elongation 74 2.50E-42 1.00E-38 ILMN_1681389 C2H2 zinc finger protein-0.40001984 INTERPRO Ubiquitin-conjugating enzyme/RWD-like 17 2.00E-05 4.20E-02 ILMN_1772218 HLA-DPA1 0.934229063 SP_PIR_KEYWORDS ribosome 60 2.00E-41 4.60E-39 ILMN_1768954 RIBC1 -0.400186083 SMART UBCc 14 1.00E-04 3.50E-02 ILMN_1778977 TYROBP 0.933302249 KEGG_PATHWAY Ribosome 65 3.80E-35 6.60E-33 ILMN_1699190 SORCS1 -0.400223681 SP_PIR_KEYWORDS ubl conjugation pathway 81 1.30E-04 2.30E-02 ILMN_1689655 HLA-DRA 0.915891173 SP_PIR_KEYWORDS protein biosynthesis 91 4.10E-34 7.20E-32 ILMN_3249088 LOC93432 -0.400285215 GOTERM_MF_FAT small conjugating protein ligase activity 35 1.40E-04 4.40E-02 ILMN_3228688 HLA-DRB1 0.906190291 SP_PIR_KEYWORDS ribonucleoprotein 114 4.80E-34 6.70E-32 ILMN_1680436 CSH2 -0.400299744 SP_PIR_KEYWORDS ligase 54 1.50E-04 2.00E-02 ILMN_2157441 HLA-DRA 0.902996561 GOTERM_CC_FAT cytosolic ribosome 59 3.20E-33 2.30E-30 ILMN_1722755 KRTAP6-2 -0.400334007 GOTERM_MF_FAT acid-amino acid ligase activity 40 1.60E-04 4.00E-02 ILMN_2066066 HLA-DRB6 0.901531942 SP_PIR_KEYWORDS -

Amyloid Precursor Protein Interaction Network in Human Testis
Silva et al. BMC Bioinformatics (2015) 16:12 DOI 10.1186/s12859-014-0432-9 RESEARCH ARTICLE Open Access Amyloid precursor protein interaction network in human testis: sentinel proteins for male reproduction Joana Vieira Silva1†, Sooyeon Yoon2†, Sara Domingues3, Sofia Guimarães3, Alexander V Goltsev2, Edgar Figueiredo da Cruz e Silva1ˆ, José Fernando F Mendes2, Odete Abreu Beirão da Cruz e Silva3 and Margarida Fardilha1,4* Abstract Background: Amyloid precursor protein (APP) is widely recognized for playing a central role in Alzheimer's disease pathogenesis. Although APP is expressed in several tissues outside the human central nervous system, the functions of APP and its family members in other tissues are still poorly understood. APP is involved in several biological functions which might be potentially important for male fertility, such as cell adhesion, cell motility, signaling, and apoptosis. Furthermore, APP superfamily members are known to be associated with fertility. Knowledge on the protein networks of APP in human testis and spermatozoa will shed light on the function of APP in the male reproductive system. Results: We performed a Yeast Two-Hybrid screen and a database search to study the interaction network of APP in human testis and sperm. To gain insights into the role of APP superfamily members in fertility, the study was extended to APP-like protein 2 (APLP2). We analyzed several topological properties of the APP interaction network and the biological and physiological properties of the proteins in the APP interaction network were also specified by gene ontologyand pathways analyses. We classified significant features related to the human male reproduction for the APP interacting proteins and identified modules of proteins with similar functional roles which may show cooperative behavior for male fertility. -

WO 2016/004387 Al 7 January 2016 (07.01.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/004387 Al 7 January 2016 (07.01.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 35/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 15/039 108 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 2 July 2015 (02.07.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 62/020,3 10 2 July 2014 (02.07.2014) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: H. LEE MOFFITT CANCER CENTER GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, AND RESEARCH INSTITUTE, INC. [US/US]; 12902 TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, Magnolia Dr., Tampa, FL 336 12-9497 (US).