Open Sadiesteffens Dissertation Final.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The VLDL Receptor Regulates Membrane Progesterone Receptor
© 2018. Published by The Company of Biologists Ltd | Journal of Cell Science (2018) 131, jcs212522. doi:10.1242/jcs.212522 RESEARCH ARTICLE The VLDL receptor regulates membrane progesterone receptor trafficking and non-genomic signaling Nancy Nader, Maya Dib, Raphael Courjaret, Rawad Hodeify, Raya Machaca, Johannes Graumann and Khaled Machaca* ABSTRACT the plasma membrane and interact with the classical P4 receptor, is Progesterone mediates its physiological functions through activation of nonetheless effective at mediating non-genomic P4 signaling both transcription-coupled nuclear receptors and seven-pass- (Bandyopadhyay et al., 1998; Dressing et al., 2011; Peluso et al., transmembrane progesterone receptors (mPRs), which transduce 2002). These results argued for the presence of membrane P4 the rapid non-genomic actions of progesterone by coupling to various receptors that are distinct from the nuclear P4 receptors. In 2003, the signaling modules. However, the immediate mechanisms of action Thomas laboratory identified a family of membrane progesterone downstream of mPRs remain in question. Herein, we use an untargeted receptors (mPRs) from fish ovaries (Zhu et al., 2003a,b) that belong quantitative proteomics approach to identify mPR interactors to better to the progestin and adiponectin (AdipoQ) receptor family (also define progesterone non-genomic signaling. Surprisingly, we identify named PAQ receptors). However, the signal transduction cascade the very-low-density lipoprotein receptor (VLDLR) as an mPRβ downstream of mPRs that mediates the non-genomic actions of P4 (PAQR8) partner that is required for mPRβ plasma membrane remains unclear. localization. Knocking down VLDLR abolishes non-genomic The non-genomic action of mPR and the ensuing signaling progesterone signaling, which is rescued by overexpressing VLDLR. -

Progesterone Receptor Membrane Component 1 Promotes Survival of Human Breast Cancer Cells and the Growth of Xenograft Tumors
Cancer Biology & Therapy ISSN: 1538-4047 (Print) 1555-8576 (Online) Journal homepage: http://www.tandfonline.com/loi/kcbt20 Progesterone receptor membrane component 1 promotes survival of human breast cancer cells and the growth of xenograft tumors Nicole C. Clark, Anne M. Friel, Cindy A. Pru, Ling Zhang, Toshi Shioda, Bo R. Rueda, John J. Peluso & James K. Pru To cite this article: Nicole C. Clark, Anne M. Friel, Cindy A. Pru, Ling Zhang, Toshi Shioda, Bo R. Rueda, John J. Peluso & James K. Pru (2016) Progesterone receptor membrane component 1 promotes survival of human breast cancer cells and the growth of xenograft tumors, Cancer Biology & Therapy, 17:3, 262-271, DOI: 10.1080/15384047.2016.1139240 To link to this article: http://dx.doi.org/10.1080/15384047.2016.1139240 Accepted author version posted online: 19 Jan 2016. Published online: 19 Jan 2016. Submit your article to this journal Article views: 49 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=kcbt20 Download by: [University of Connecticut] Date: 26 May 2016, At: 11:28 CANCER BIOLOGY & THERAPY 2016, VOL. 17, NO. 3, 262–271 http://dx.doi.org/10.1080/15384047.2016.1139240 RESEARCH PAPER Progesterone receptor membrane component 1 promotes survival of human breast cancer cells and the growth of xenograft tumors Nicole C. Clarka,*, Anne M. Frielb,*, Cindy A. Prua, Ling Zhangb, Toshi Shiodac, Bo R. Ruedab, John J. Pelusod, and James K. Prua aDepartment of Animal Sciences, -

CST9L (NM 080610) Human Tagged ORF Clone – RC206646L4
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC206646L4 CST9L (NM_080610) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: CST9L (NM_080610) Human Tagged ORF Clone Tag: mGFP Symbol: CST9L Synonyms: bA218C14.1; CTES7B Vector: pLenti-C-mGFP-P2A-Puro (PS100093) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: Puromycin ORF Nucleotide The ORF insert of this clone is exactly the same as(RC206646). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_080610 ORF Size: 441 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 CST9L (NM_080610) Human Tagged ORF Clone – RC206646L4 OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_080610.1 RefSeq Size: 982 bp RefSeq ORF: 444 bp Locus ID: 128821 UniProt ID: Q9H4G1, A0A140VJH1 Protein Families: Secreted Protein, Transmembrane MW: 17.3 kDa Gene Summary: The cystatin superfamily encompasses proteins that contain multiple cystatin-like sequences. -

In Silico Analysis Identifies Genes Common Between Five Primary Gastrointestinal Cancer Sites with Potential Clinical Applications
ORIGINAL ARTICLE Annals of Gastroenterology (2014) 27, 1-14 In silico analysis identifies genes common between five primary gastrointestinal cancer sites with potential clinical applications Subhankar Chakraborty University of Nebraska Medical Center, Omaha, NE, USA Abstract Background Previous studies have investigated differential gene expression in gastrointes- tinal (GI) epithelial cancers by microarray. The aim of the present study was to use data from the Oncomine database to identify genes that share a similar differential expression in two or more primary GI cancer sites. Methods Five thousand of the most differentially expressed genes in epithelial cancers (com- pared to normal tissue) arising in the pancreas, liver, stomach, esophagus or colorectum were identified (1,000 per primary site) from Oncomine. Using Venn diagrams, genes common to two or more primary GI sites were identified. Functional and pathway analysis was performed on genes that were similarly expressed in ≥3 of the five areas of the GI tract. Results Forty six studies comprising 5,876 samples were included. Overall, 90.6% genes were unique to the respective primary sites, 7.4% shared between two GI primary sites, 1.8% between three and 0.2% between four GI primary sites. Pancreatic and hepatocellular cancers (HCC) shared most number of upregulated genes (N=66) while HCC and gastric cancer shared most downregulated genes (N=59). Genes encoding enzymes comprised the most commonly shared genes between GI primary sites (30.4% of upregulated and 63.2% of downregulated genes). Those genes that were shared between three or more GI primary sites also showed significant differential expression in the same direction in other non-GI cancers. -

The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc Oncogenesis
The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis By Yuting Sun This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy at the University of New South Wales Children’s Cancer Institute Australia for Medical Research School of Women’s and Children’s Health, Faculty of Medicine University of New South Wales Australia August 2014 PLEASE TYPE THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Sun First name: Yuting Other name/s: Abbreviation for degree as given in the University calendar: PhD School : School of·Women's and Children's Health Faculty: Faculty of Medicine Title: The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis. Abstract 350 words maximum: (PLEASE TYPE) N-Myc Induces neuroblastoma by regulating the expression of target genes and proteins, and N-Myc protein is degraded by Fbxw7 and NEDD4 and stabilized by Aurora A. The class lla histone deacetylase HDAC5 suppresses gene transcription, and blocks myoblast and leukaemia cell differentiation. While histone H3 lysine 4 (H3K4) trimethylation at target gene promoters is a pre-requisite for Myc· induced transcriptional activation, WDRS, as a histone H3K4 methyltransferase presenter, is required for H3K4 methylation and transcriptional activation mediated by a histone H3K4 methyltransferase complex. Here, I investigated the roles of HDAC5 and WDR5 in N-Myc overexpressing neuroblastoma. I have found that N-Myc upregulates HDAC5 protein expression, and that HDAC5 represses NEDD4 gene expression, increases Aurora A gene expression and consequently upregulates N-Myc protein expression in neuroblastoma cells. -

Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis
Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma, Anna-Karin Ekman, Cecilia Bivik Eding and Charlotta Enerbäck The self-archived postprint version of this journal article is available at Linköping University Institutional Repository (DiVA): http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-147791 N.B.: When citing this work, cite the original publication. Verma, D., Ekman, A., Bivik Eding, C., Enerbäck, C., (2018), Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis, Journal of Investigative Dermatology, 138(5), 1088-1093. https://doi.org/10.1016/j.jid.2017.11.036 Original publication available at: https://doi.org/10.1016/j.jid.2017.11.036 Copyright: Elsevier http://www.elsevier.com/ Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma*a, Anna-Karin Ekman*a, Cecilia Bivik Edinga and Charlotta Enerbäcka *Authors contributed equally aIngrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Division of Dermatology, Linköping University, Linköping, Sweden Corresponding author: Charlotta Enerbäck Ingrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Linköping University SE-581 85 Linköping, Sweden Phone: +46 10 103 7429 E-mail: [email protected] Short title Differential methylation in psoriasis Abbreviations CGI, CpG island; DMS, differentially methylated site; RRBS, reduced representation bisulphite sequencing Keywords (max 6) psoriasis, epidermis, methylation, Wnt, susceptibility, expression 1 ABSTRACT Psoriasis is a chronic inflammatory skin disease with both local and systemic components. Genome-wide approaches have identified more than 60 psoriasis-susceptibility loci, but genes are estimated to explain only one third of the heritability in psoriasis, suggesting additional, yet unidentified, sources of heritability. -

Understanding Chronic Kidney Disease: Genetic and Epigenetic Approaches
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Understanding Chronic Kidney Disease: Genetic And Epigenetic Approaches Yi-An Ko Ko University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, Genetics Commons, and the Systems Biology Commons Recommended Citation Ko, Yi-An Ko, "Understanding Chronic Kidney Disease: Genetic And Epigenetic Approaches" (2017). Publicly Accessible Penn Dissertations. 2404. https://repository.upenn.edu/edissertations/2404 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2404 For more information, please contact [email protected]. Understanding Chronic Kidney Disease: Genetic And Epigenetic Approaches Abstract The work described in this dissertation aimed to better understand the genetic and epigenetic factors influencing chronic kidney disease (CKD) development. Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) significantly associated with chronic kidney disease. However, these studies have not effectively identified target genes for the CKD variants. Most of the identified variants are localized to non-coding genomic regions, and how they associate with CKD development is not well-understood. As GWAS studies only explain a small fraction of heritability, we hypothesized that epigenetic changes could explain part of this missing heritability. To identify potential gene targets of the genetic variants, we performed expression quantitative loci (eQTL) analysis, using genotyping arrays and RNA sequencing from human kidney samples. To identify the target genes of CKD-associated SNPs, we integrated the GWAS-identified SNPs with the eQTL results using a Bayesian colocalization method, coloc. This resulted in a short list of target genes, including PGAP3 and CASP9, two genes that have been shown to present with kidney phenotypes in knockout mice. -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Progesterone – Friend Or Foe?
Frontiers in Neuroendocrinology 59 (2020) 100856 Contents lists available at ScienceDirect Frontiers in Neuroendocrinology journal homepage: www.elsevier.com/locate/yfrne Progesterone – Friend or foe? T ⁎ Inger Sundström-Poromaaa, , Erika Comascob, Rachael Sumnerc, Eileen Ludersd,e a Department of Women’s and Children’s Health, Uppsala University, Sweden b Department of Neuroscience, Science for Life Laboratory, Uppsala University, Uppsala, Sweden c School of Pharmacy, University of Auckland, New Zealand d School of Psychology, University of Auckland, New Zealand e Laboratory of Neuro Imaging, School of Medicine, University of Southern California, Los Angeles, USA ARTICLE INFO ABSTRACT Keywords: Estradiol is the “prototypic” sex hormone of women. Yet, women have another sex hormone, which is often Allopregnanolone disregarded: Progesterone. The goal of this article is to provide a comprehensive review on progesterone, and its Emotion metabolite allopregnanolone, emphasizing three key areas: biological properties, main functions, and effects on Hormonal contraceptives mood in women. Recent years of intensive research on progesterone and allopregnanolone have paved the way Postpartum depression for new treatment of postpartum depression. However, treatment for premenstrual syndrome and premenstrual Premenstrual dysphoric disorder dysphoric disorder as well as contraception that women can use without risking mental health problems are still Progesterone needed. As far as progesterone is concerned, we might be dealing with a two-edged sword: while its metabolite allopregnanolone has been proven useful for treatment of PPD, it may trigger negative symptoms in women with PMS and PMDD. Overall, our current knowledge on the beneficial and harmful effects of progesterone is limited and further research is imperative. Introduction 1. -
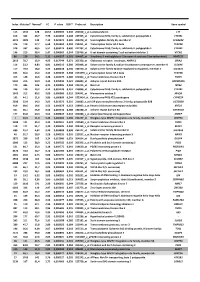
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244 -

Human CST9L / Testatin Protein (Fc Tag)
Human CST9L / Testatin Protein (Fc Tag) Catalog Number: 13243-H02H General Information SDS-PAGE: Gene Name Synonym: bA218C14.1; CTES7B; PRO3543; UNQ1835 Protein Construction: A DNA sequence encoding the human CST9L (Q9H4G1) (Met 1-His 147) was fused with the Fc region of human IgG1 at the C-terminus. Source: Human Expression Host: Human Cells QC Testing Purity: > 92 % as determined by SDS-PAGE Endotoxin: Protein Description < 1.0 EU per μg of the protein as determined by the LAL method Testatin is a member of the Cystatin family. Cystatins comprise genes that Stability: all show expression patterns that are strikingly restricted to reproductive tissue. Cystatins are a family of cysteine protease inhibitors with homology Samples are stable for up to twelve months from date of receipt at -70 ℃ to chicken cystatin. There are typically about 115 amino acids in this family. They are largely acidic, contain four conserved cysteine residues known to Predicted N terminal: Trp 29 form two disulfide bonds, may be glycosylated and/or phosphorylated, with Molecular Mass: similarity to fetuins, kininogens, stefins, histidine-rich glycoproteins and cystatin-related proteins. Testatin shows homology to family 2 cystatins, a The recombinant human CST9L/Fc chimera is a disulfide-linked group of broadly expressed small secretory proteins that are inhibitors of homodimeric protein. The reduced monomer consists of 360 amino acids cysteine proteases in vitro but whose in vivo functions are unclear. It is and has a calculated molecular mass of 41.3 kDa. In SDS-PAGE under expressed in germ cells and somatic cells in reproductive tissues. Testatin reducing conditions, the apparent molecular mass of rhCST9L/Fc is considered a strong candidate for involvement in early testis monomer is approximately 48 kDa.