Amyloid Precursor Protein Interaction Network in Human Testis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

I HIGH MASS ACCURACY COUPLED to SPATIALLY-DIRECTED
HIGH MASS ACCURACY COUPLED TO SPATIALLY-DIRECTED PROTEOMICS FOR IMPROVED PROTEIN IDENTIFICATIONS IN IMAGING MASS SPECTROMETRY EXPERIMENTS By David Geoffrey Rizzo Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Chemistry August, 2016 Nashville, Tennessee Approved: Richard M. Caprioli, Ph.D. Kevin L. Schey, Ph.D. John A. McLean, Ph.D. Michael P. Stone, Ph.D. i Copyright © 2016 by David Geoffrey Rizzo All Rights Reserved ii This work is dedicated to my family and friends, who have shown nothing but support for me in all of life’s endeavors. iii ACKNOWLEDGEMENTS “As we express our gratitude, we must never forget that the highest appreciation is not to utter words, but to live by them.” - John F. Kennedy – There are many people I must thank for showing kindness, encouragement, and support for me during my tenure as a graduate student. First and foremost, I would like to thank my research advisor, Richard Caprioli, for providing both ample resources and guidance that allowed me to grow as a scientist. Our discussions about my research and science in general have helped me become a much more focused and discerning analytical chemist. I must also thank my Ph.D. committee members, Drs. Kevin Schey, John McLean, and Michael Stone, who have brought valuable insight into my research and provided direction along the way. My undergraduate advisor, Dr. Facundo Fernández, encouraged me to begin research in his lab and introduced me to the world of mass spectrometry. -

In Silico Analysis Identifies Genes Common Between Five Primary Gastrointestinal Cancer Sites with Potential Clinical Applications
ORIGINAL ARTICLE Annals of Gastroenterology (2014) 27, 1-14 In silico analysis identifies genes common between five primary gastrointestinal cancer sites with potential clinical applications Subhankar Chakraborty University of Nebraska Medical Center, Omaha, NE, USA Abstract Background Previous studies have investigated differential gene expression in gastrointes- tinal (GI) epithelial cancers by microarray. The aim of the present study was to use data from the Oncomine database to identify genes that share a similar differential expression in two or more primary GI cancer sites. Methods Five thousand of the most differentially expressed genes in epithelial cancers (com- pared to normal tissue) arising in the pancreas, liver, stomach, esophagus or colorectum were identified (1,000 per primary site) from Oncomine. Using Venn diagrams, genes common to two or more primary GI sites were identified. Functional and pathway analysis was performed on genes that were similarly expressed in ≥3 of the five areas of the GI tract. Results Forty six studies comprising 5,876 samples were included. Overall, 90.6% genes were unique to the respective primary sites, 7.4% shared between two GI primary sites, 1.8% between three and 0.2% between four GI primary sites. Pancreatic and hepatocellular cancers (HCC) shared most number of upregulated genes (N=66) while HCC and gastric cancer shared most downregulated genes (N=59). Genes encoding enzymes comprised the most commonly shared genes between GI primary sites (30.4% of upregulated and 63.2% of downregulated genes). Those genes that were shared between three or more GI primary sites also showed significant differential expression in the same direction in other non-GI cancers. -

Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis
Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma, Anna-Karin Ekman, Cecilia Bivik Eding and Charlotta Enerbäck The self-archived postprint version of this journal article is available at Linköping University Institutional Repository (DiVA): http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-147791 N.B.: When citing this work, cite the original publication. Verma, D., Ekman, A., Bivik Eding, C., Enerbäck, C., (2018), Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis, Journal of Investigative Dermatology, 138(5), 1088-1093. https://doi.org/10.1016/j.jid.2017.11.036 Original publication available at: https://doi.org/10.1016/j.jid.2017.11.036 Copyright: Elsevier http://www.elsevier.com/ Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma*a, Anna-Karin Ekman*a, Cecilia Bivik Edinga and Charlotta Enerbäcka *Authors contributed equally aIngrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Division of Dermatology, Linköping University, Linköping, Sweden Corresponding author: Charlotta Enerbäck Ingrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Linköping University SE-581 85 Linköping, Sweden Phone: +46 10 103 7429 E-mail: [email protected] Short title Differential methylation in psoriasis Abbreviations CGI, CpG island; DMS, differentially methylated site; RRBS, reduced representation bisulphite sequencing Keywords (max 6) psoriasis, epidermis, methylation, Wnt, susceptibility, expression 1 ABSTRACT Psoriasis is a chronic inflammatory skin disease with both local and systemic components. Genome-wide approaches have identified more than 60 psoriasis-susceptibility loci, but genes are estimated to explain only one third of the heritability in psoriasis, suggesting additional, yet unidentified, sources of heritability. -

Using Evolutionary Conserved Modules in Gene Networks As a Strategy to Leverage High Throughput Gene Expression Queries Jeanne M
Ecology, Evolution and Organismal Biology Ecology, Evolution and Organismal Biology Publications 9-2010 Using Evolutionary Conserved Modules in Gene Networks as a Strategy to Leverage High Throughput Gene Expression Queries Jeanne M. Serb Iowa State University, [email protected] Megan C. Orr Iowa State University M. Heather West Greenlee Iowa State University, [email protected] Follow this and additional works at: http://lib.dr.iastate.edu/eeob_ag_pubs Part of the Ecology and Evolutionary Biology Commons, Genetics Commons, and the Systems Biology Commons The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ eeob_ag_pubs/19. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Ecology, Evolution and Organismal Biology at Iowa State University Digital Repository. It has been accepted for inclusion in Ecology, Evolution and Organismal Biology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Using Evolutionary Conserved Modules in Gene Networks as a Strategy to Leverage High Throughput Gene Expression Queries Abstract Background: Large-scale gene expression studies have not yielded the expected insight into genetic networks that control complex processes. These anticipated discoveries have been limited not by technology, but by a lack of effective strategies to investigate the data in a manageable and meaningful way. Previous work suggests that using a pre-determined seednetwork of gene relationships to query large-scale expression datasets is an effective way to generate candidate genes for further study and network expansion or enrichment. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

A Potential Role for APLP2 in Axogenesis
The Journal of Neuroscience, October 1995, 15(10): 63144326 Distribution of an APP Homolog, APLP2, in the Mouse Olfactory System: A Potential Role for APLP2 in Axogenesis Gopal Thinakaran,1,5 Cheryl A. Kitt, 1,5 A. Jane I. Roskams,” Hilda H. Slunt,1,5 Eliezer Masliah,’ Cornelia von Koch,2,5 Stephen D. Ginsberg,i,5 Gabriele V. Ronnett,‘s4 Randall Ft. Reed,233I6 Donald L. Price,‘,2.4,5 and Sangram S. Sisodiaiz5 ‘Departments of Pathology, 2Neuroscience, 3Molecular Biology and Genetics, and 4Neurology, 5Neuropathology Laboratory, and the 6Howard Hughes Medical Institute, The Johns Hopkins University School of Medicine, Baltimore, Maryland, 21205 and ‘Departments of Neurosciences and Pathology and Neuroscience, University of California at San Diego, La Jolla, California 92093 Deposition of l3-amyloid (AS) in senile plaques is a major APLP2, alternative splicing, chondroitin sulfate glycosami- pathological characteristic of Alzheimer’s disease. Ap is noglycan, glomeruli, olfactory epithelium, o/factory sen- generated by proteolytic processing of amyloici precursor sory neurons] proteins (APP). APP is a member of a family of related poly- peptides that includes amyloid precursor-like proteins A principal pathological feature of Alzheimer’s disease is the APLPl and APLPS. To examine the distribution of APLP2 deposition of Al3 in brain parenchyma (Glenner and Wong, in the nervous system, we generated antibodies specific 1984; Masters et al., 1985). Al3, an -4 kDa polypeptide, is for APLPS and used these reagents in immunocytochemi- derived from larger type I integral membrane glycoproteins, cal and biochemical studies of the rodent nervous system. termed the amyloid precursor proteins (APP) (Kang et al., 1987; In this report, we document that in cortex and hippocam- Kitaguchi et al., 1988; Ponte et al., 1988; Tanzi et al., 1988; pus, APLPS is enriched in postsynaptic compartments. -

APLP2 Regulates Refractive Error and Myopia Development in Mice and Humans
Tkatchenko, A. V., Tkatchenko, T. V., Guggenheim, J. A., Verhoeven, V. J. M., Hysi, P. G., Wojciechowski, R., Singh, P. K., Kumar, A., Thinakaran, G., Williams, C. (2015). APLP2 Regulates Refractive Error and Myopia Development in Mice and Humans. PLoS Genetics, 11(8), [e1005432]. https://doi.org/10.1371/journal.pgen.1005432 Publisher's PDF, also known as Version of record License (if available): CC BY Link to published version (if available): 10.1371/journal.pgen.1005432 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Public Library of Science at http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1005432. Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/red/research-policy/pure/user-guides/ebr-terms/ RESEARCH ARTICLE APLP2 Regulates Refractive Error and Myopia Development in Mice and Humans Andrei V. Tkatchenko1,2☯*, Tatiana V. Tkatchenko1☯, Jeremy A. Guggenheim3☯, Virginie J. M. Verhoeven4,5, Pirro G. Hysi6, Robert Wojciechowski7,8, Pawan Kumar Singh9, Ashok Kumar9,10, Gopal Thinakaran11, Consortium for Refractive Error and Myopia (CREAM)¶, Cathy Williams12 1 Department of Ophthalmology, Columbia University, New York, New York, United -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
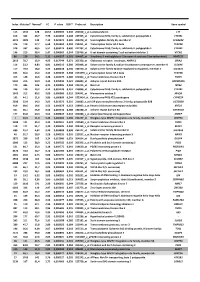
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244 -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Phénotype Des Écotypes De Touladi Du Lac Supérieur
Génomique des populations et association génotype- phénotype des écotypes de touladi du lac Supérieur Mémoire Alysse Perreault-Payette Maîtrise en biologie Maître ès sciences (M. Sc.) Québec, Canada © Alysse Perreault-Payette, 2016 Génomique des populations et association génotype- phénotype des écotypes de touladi du lac Supérieur Mémoire Alysse Perreault-Payette Sous la direction de : Louis Bernatchez, directeur de recherche Pascal Sirois, codirecteur de recherche Résumé L’apparition et le maintien d’écotypes adaptés à différentes niches écologiques, en situation de sympatrie, est régit par une multitude de facteurs. Ceux-ci sont essentiels pour la compréhension des processus évolutifs impliqués mais aussi pour la gestion et la conservation des populations en question. Le touladi (Salvelinus namaycush) est un salmonidé reconnu pour la présence d’écotypes liée à l’utilisation des ressources et de l’habitat à travers l’Amérique du Nord. Un total de quatre écotypes a été décrit vivant dans le lac Supérieur, se différenciant par l’habitat utilisé, l’alimentation, la morphologie ainsi que l’ostéologie. L’objectif principal de la présente étude était de quantifier l’étendue de la différentiation génétique entre les différents sites d’échantillonnage ainsi qu’entre les différents écotypes. Un second objectif était d’identifier des marqueurs potentiellement sous sélection entre les différents écotypes reflétant de possibles adaptations locales. Pour ce faire, un total de 486 individus, représentant les quatre écotypes pour chacun des quatre sites d’échantillonnages, a été génotypé à 6822 SNPs (polymorphisme de nucléotide simple). De plus, des analyses morphométriques ont été effectuées afin de caractériser l’ampleur de la divergence morphologique entre les écotypes à chacun des sites. -

Supplementary Information.Pdf
Supplementary Information Whole transcriptome profiling reveals major cell types in the cellular immune response against acute and chronic active Epstein‐Barr virus infection Huaqing Zhong1, Xinran Hu2, Andrew B. Janowski2, Gregory A. Storch2, Liyun Su1, Lingfeng Cao1, Jinsheng Yu3, and Jin Xu1 Department of Clinical Laboratory1, Children's Hospital of Fudan University, Minhang District, Shanghai 201102, China; Departments of Pediatrics2 and Genetics3, Washington University School of Medicine, Saint Louis, Missouri 63110, United States. Supplementary information includes the following: 1. Supplementary Figure S1: Fold‐change and correlation data for hyperactive and hypoactive genes. 2. Supplementary Table S1: Clinical data and EBV lab results for 110 study subjects. 3. Supplementary Table S2: Differentially expressed genes between AIM vs. Healthy controls. 4. Supplementary Table S3: Differentially expressed genes between CAEBV vs. Healthy controls. 5. Supplementary Table S4: Fold‐change data for 303 immune mediators. 6. Supplementary Table S5: Primers used in qPCR assays. Supplementary Figure S1. Fold‐change (a) and Pearson correlation data (b) for 10 cell markers and 61 hypoactive and hyperactive genes identified in subjects with acute EBV infection (AIM) in the primary cohort. Note: 23 up‐regulated hyperactive genes were highly correlated positively with cytotoxic T cell (Tc) marker CD8A and NK cell marker CD94 (KLRD1), and 38 down‐regulated hypoactive genes were highly correlated positively with B cell, conventional dendritic cell