Cyclin D1 and CDK4 Activity Contribute to the Undifferentiated Phenotype in Neuroblastoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In Silico Analysis Identifies Genes Common Between Five Primary Gastrointestinal Cancer Sites with Potential Clinical Applications
ORIGINAL ARTICLE Annals of Gastroenterology (2014) 27, 1-14 In silico analysis identifies genes common between five primary gastrointestinal cancer sites with potential clinical applications Subhankar Chakraborty University of Nebraska Medical Center, Omaha, NE, USA Abstract Background Previous studies have investigated differential gene expression in gastrointes- tinal (GI) epithelial cancers by microarray. The aim of the present study was to use data from the Oncomine database to identify genes that share a similar differential expression in two or more primary GI cancer sites. Methods Five thousand of the most differentially expressed genes in epithelial cancers (com- pared to normal tissue) arising in the pancreas, liver, stomach, esophagus or colorectum were identified (1,000 per primary site) from Oncomine. Using Venn diagrams, genes common to two or more primary GI sites were identified. Functional and pathway analysis was performed on genes that were similarly expressed in ≥3 of the five areas of the GI tract. Results Forty six studies comprising 5,876 samples were included. Overall, 90.6% genes were unique to the respective primary sites, 7.4% shared between two GI primary sites, 1.8% between three and 0.2% between four GI primary sites. Pancreatic and hepatocellular cancers (HCC) shared most number of upregulated genes (N=66) while HCC and gastric cancer shared most downregulated genes (N=59). Genes encoding enzymes comprised the most commonly shared genes between GI primary sites (30.4% of upregulated and 63.2% of downregulated genes). Those genes that were shared between three or more GI primary sites also showed significant differential expression in the same direction in other non-GI cancers. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis
Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma, Anna-Karin Ekman, Cecilia Bivik Eding and Charlotta Enerbäck The self-archived postprint version of this journal article is available at Linköping University Institutional Repository (DiVA): http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-147791 N.B.: When citing this work, cite the original publication. Verma, D., Ekman, A., Bivik Eding, C., Enerbäck, C., (2018), Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis, Journal of Investigative Dermatology, 138(5), 1088-1093. https://doi.org/10.1016/j.jid.2017.11.036 Original publication available at: https://doi.org/10.1016/j.jid.2017.11.036 Copyright: Elsevier http://www.elsevier.com/ Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma*a, Anna-Karin Ekman*a, Cecilia Bivik Edinga and Charlotta Enerbäcka *Authors contributed equally aIngrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Division of Dermatology, Linköping University, Linköping, Sweden Corresponding author: Charlotta Enerbäck Ingrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Linköping University SE-581 85 Linköping, Sweden Phone: +46 10 103 7429 E-mail: [email protected] Short title Differential methylation in psoriasis Abbreviations CGI, CpG island; DMS, differentially methylated site; RRBS, reduced representation bisulphite sequencing Keywords (max 6) psoriasis, epidermis, methylation, Wnt, susceptibility, expression 1 ABSTRACT Psoriasis is a chronic inflammatory skin disease with both local and systemic components. Genome-wide approaches have identified more than 60 psoriasis-susceptibility loci, but genes are estimated to explain only one third of the heritability in psoriasis, suggesting additional, yet unidentified, sources of heritability. -

Figure S1. Representative Report Generated by the Ion Torrent System Server for Each of the KCC71 Panel Analysis and Pcafusion Analysis
Figure S1. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. A Figure S1. Continued. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. B Figure S2. Comparative analysis of the variant frequency found by the KCC71 panel and calculated from publicly available cBioPortal datasets. For each of the 71 genes in the KCC71 panel, the frequency of variants was calculated as the variant number found in the examined cases. Datasets marked with different colors and sample numbers of prostate cancer are presented in the upper right. *Significantly high in the present study. Figure S3. Seven subnetworks extracted from each of seven public prostate cancer gene networks in TCNG (Table SVI). Blue dots represent genes that include initial seed genes (parent nodes), and parent‑child and child‑grandchild genes in the network. Graphical representation of node‑to‑node associations and subnetwork structures that differed among and were unique to each of the seven subnetworks. TCNG, The Cancer Network Galaxy. Figure S4. REVIGO tree map showing the predicted biological processes of prostate cancer in the Japanese. Each rectangle represents a biological function in terms of a Gene Ontology (GO) term, with the size adjusted to represent the P‑value of the GO term in the underlying GO term database. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -
Animal Cells Anterior Epidermis Anterior Epidermis A-Neural
Anterior Epidermis Anterior Epidermis KH2012 TF common name Log2FC -Log(pvalue) Log2FC -Log(pvalue) DE in Imai Matched PWM PWM Cluster KH2012 TF common name Log2FC -Log(pvalue) Log2FC -Log(pvalue) DE in Imai Matched PWM PWM Cluster gene model Sibling Cluster Sibling Cluster Parent Cluster Parent Cluster z-score z-score gene model Sibling Cluster Sibling Cluster Parent Cluster Parent Cluster z-score z-score KH2012:KH.C11.485 Irx-B 1.0982 127.5106 0.9210 342.5323 Yes No PWM Hits No PWM Hits KH2012:KH.C1.159 E(spl)/hairy-a 1.3445 65.6302 0.6908 14.3413 Not Analyzed -15.2125 No PWM Hits KH2012:KH.L39.1 FoxH-b 0.6677 45.8148 1.1074 185.2909 No -3.3335 5.2695 KH2012:KH.C1.99 SoxB1 1.2482 73.2413 0.3331 9.3534 Not Analyzed No PWM match No PWM match KH2012:KH.C1.159 E(spl)/hairy-a 0.6233 47.2239 0.4339 77.0192 Yes -10.496 No PWM Hits KH2012:KH.C11.485 Irx-B 1.2355 72.8859 0.1608 0.0137 Not Analyzed No PWM Hits No PWM Hits KH2012:KH.C7.43 AP-2-like2 0.4991 31.7939 0.4775 68.8091 Yes 10.551 21.586 KH2012:KH.C1.1016 GCNF 0.8556 36.2030 1.3828 100.1236 Not Analyzed No PWM match No PWM match KH2012:KH.C1.99 SoxB1 0.4913 33.7808 0.3406 39.0890 No No PWM match No PWM match KH2012:KH.L108.4 CREB/ATF-a 0.6859 37.8207 0.3453 15.8154 Not Analyzed 6.405 8.6245 KH2012:KH.C7.157 Emc 0.4139 19.2080 1.1001 173.3024 Yes No PWM match No PWM match KH2012:KH.S164.12 SoxB2 0.6194 22.8414 0.6433 35.7335 Not Analyzed 8.722 17.405 KH2012:KH.C4.366 ERF2 -0.4878 -32.3767 -0.1770 -0.2316 No -10.324 9.7885 KH2012:KH.L4.17 Zinc Finger (C2H2)-18 0.6166 24.8925 0.2386 5.3130 -

MTGR1 (CBFA2T2) (NM 001039709) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG202013 MTGR1 (CBFA2T2) (NM_001039709) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: MTGR1 (CBFA2T2) (NM_001039709) Human Tagged ORF Clone Tag: TurboGFP Symbol: CBFA2T2 Synonyms: EHT; MTGR1; p85; ZMYND3 Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 MTGR1 (CBFA2T2) (NM_001039709) Human Tagged ORF Clone – RG202013 ORF Nucleotide >RG202013 representing NM_001039709 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGGGTTTCACCATGTTGGCCAGGCTCGTCTTGAACTCCTGACCTCAGGTGATCTGCCTGCATTGGCCT CCCAACGTGCTGGGATTACAGTTGGTCCTGAGAAAAGGGTGCCAGCGATGCCTGGATCGCCTGTGGAAGT GAAGATACAGTCCAGATCCTCACCTCCCACCATGCCACCCCTCCCACCAATAAATCCTGGAGGACCGAGG CCAGTGTCCTTCACTCCTACTGCATTAAGCAATGGCATCAACCATTCTCCTCCTACCCTGAATGGTGCCC CATCACCGCCACAGAGATTCAGCAATGGTCCTGCCTCCTCCACATCATCTGCACTCACAAATCAGCAATT GCCAGCCACTTGTGGTGCTCGACAACTCAGCAAGTTGAAACGCTTTCTTACCACTCTGCAACAGTTTGGC AATGACATCTCCCCTGAGATTGGGGAGAAGGTGCGGACTCTTGTTCTTGCACTGGTGAACTCAACAGTGA CAATTGAGGAATTCCACTGTAAGCTCCAAGAAGCCACAAACTTTCCCCTTCGTCCTTTTGTGATTCCATT -

Methylome and Transcriptome Maps of Human Visceral and Subcutaneous
www.nature.com/scientificreports OPEN Methylome and transcriptome maps of human visceral and subcutaneous adipocytes reveal Received: 9 April 2019 Accepted: 11 June 2019 key epigenetic diferences at Published: xx xx xxxx developmental genes Stephen T. Bradford1,2,3, Shalima S. Nair1,3, Aaron L. Statham1, Susan J. van Dijk2, Timothy J. Peters 1,3,4, Firoz Anwar 2, Hugh J. French 1, Julius Z. H. von Martels1, Brodie Sutclife2, Madhavi P. Maddugoda1, Michelle Peranec1, Hilal Varinli1,2,5, Rosanna Arnoldy1, Michael Buckley1,4, Jason P. Ross2, Elena Zotenko1,3, Jenny Z. Song1, Clare Stirzaker1,3, Denis C. Bauer2, Wenjia Qu1, Michael M. Swarbrick6, Helen L. Lutgers1,7, Reginald V. Lord8, Katherine Samaras9,10, Peter L. Molloy 2 & Susan J. Clark 1,3 Adipocytes support key metabolic and endocrine functions of adipose tissue. Lipid is stored in two major classes of depots, namely visceral adipose (VA) and subcutaneous adipose (SA) depots. Increased visceral adiposity is associated with adverse health outcomes, whereas the impact of SA tissue is relatively metabolically benign. The precise molecular features associated with the functional diferences between the adipose depots are still not well understood. Here, we characterised transcriptomes and methylomes of isolated adipocytes from matched SA and VA tissues of individuals with normal BMI to identify epigenetic diferences and their contribution to cell type and depot-specifc function. We found that DNA methylomes were notably distinct between diferent adipocyte depots and were associated with diferential gene expression within pathways fundamental to adipocyte function. Most striking diferential methylation was found at transcription factor and developmental genes. Our fndings highlight the importance of developmental origins in the function of diferent fat depots. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -
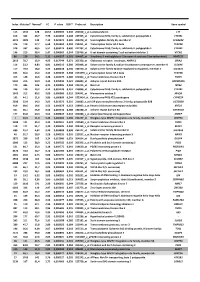
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244 -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

A Pathologic Link Between Wilms Tumor Suppressor Gene, WT1, And
Volume 10 Number 1 January 2008 pp. 69–78 69 www.neoplasia.com RESEARCH ARTICLE † Marianne K.-H. Kim*, Jacqueline M. Mason , A Pathologic Link between Wilms ‡ § Chi-Ming Li , Windy Berkofsky-Fessler , ∥ WT1 Le Jiang , Divaker Choubey¶, Paul E. Grundy#, Tumor Suppressor Gene, , ∥ and IFI161,2 Benjamin Tycko and Jonathan D. Licht* *Division of Hematology/Oncology, Feinberg School of Medicine, Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL, USA; †The Campbell Family Institute for Breast Cancer Research at the Ontario, Cancer Institute, Ontario, Canada; ‡Translational Medicine, Amgen, Thousand Oaks, CA, USA; §Section of Bioinformatics, Genetics and Genomics, Hoffmann-La Roche Inc, Nutley, NJ, USA; ∥Institute for Cancer Genetics and Department of Pathology, Columbia University College of Physicians and Surgeons, New York, NY, USA; ¶University of Cincinnati, Cincinnati, OH, USA; #University of Alberta, Alberta, Canada Abstract The Wilms tumor gene (WT1) is mutated or deleted in patients with heredofamilial syndromes associated with the development of Wilms tumors, but is infrequently mutated in sporadic Wilms tumors. By comparing the micro- array profiles of syndromic versus sporadic Wilms tumors and WT1-inducible Saos-2 osteosarcoma cells, we iden- tified interferon-inducible protein 16 (IFI16), a transcriptional modulator, as a differentially expressed gene and a candidate WT1 target gene. WT1 induction in Saos-2 osteosarcoma cells led to strong induction of IFI16 expression and its promoter activity was responsive to the WT1 protein. Immunohistochemical analysis showed that IFI16 and WT1 colocalized in WT1-replete Wilms tumors, but not in normal human midgestation fetal kidneys, suggesting that the ability of WT1 to regulate IFI16 in tumors represented an aberrant pathologic relationship.