Biochimica Et Biophysica Acta 1863 (2016) 1307–1318
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Essentiality Landscape and Druggable Oncogenic Dependencies in Herpesviral Primary Effusion Lymphoma
ARTICLE DOI: 10.1038/s41467-018-05506-9 OPEN Gene essentiality landscape and druggable oncogenic dependencies in herpesviral primary effusion lymphoma Mark Manzano1, Ajinkya Patil1, Alexander Waldrop2, Sandeep S. Dave2, Amir Behdad3 & Eva Gottwein1 Primary effusion lymphoma (PEL) is caused by Kaposi’s sarcoma-associated herpesvirus. Our understanding of PEL is poor and therefore treatment strategies are lacking. To address this 1234567890():,; need, we conducted genome-wide CRISPR/Cas9 knockout screens in eight PEL cell lines. Integration with data from unrelated cancers identifies 210 genes as PEL-specific oncogenic dependencies. Genetic requirements of PEL cell lines are largely independent of Epstein-Barr virus co-infection. Genes of the NF-κB pathway are individually non-essential. Instead, we demonstrate requirements for IRF4 and MDM2. PEL cell lines depend on cellular cyclin D2 and c-FLIP despite expression of viral homologs. Moreover, PEL cell lines are addicted to high levels of MCL1 expression, which are also evident in PEL tumors. Strong dependencies on cyclin D2 and MCL1 render PEL cell lines highly sensitive to palbociclib and S63845. In summary, this work comprehensively identifies genetic dependencies in PEL cell lines and identifies novel strategies for therapeutic intervention. 1 Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. 2 Duke Cancer Institute and Center for Genomic and Computational Biology, Duke University, Durham, NC 27708, USA. 3 Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. Correspondence and requests for materials should be addressed to E.G. (email: [email protected]) NATURE COMMUNICATIONS | (2018) 9:3263 | DOI: 10.1038/s41467-018-05506-9 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-05506-9 he human oncogenic γ-herpesvirus Kaposi’s sarcoma- (IRF4), a critical oncogene in multiple myeloma33. -

Large-Scale Image-Based Profiling of Single-Cell Phenotypes in Arrayed CRISPR-Cas9 Gene Perturbation Screens
Published online: January 23, 2018 Method Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR-Cas9 gene perturbation screens Reinoud de Groot1 , Joel Lüthi1,2 , Helen Lindsay1 , René Holtackers1 & Lucas Pelkmans1,* Abstract this reason, CRISPR-Cas9 has been used in large-scale functional genomic screens (Shalem et al, 2015). Most screens performed to High-content imaging using automated microscopy and computer date employ a pooled screening strategy, which can identify genes vision allows multivariate profiling of single-cell phenotypes. Here, that cause differential growth in screening conditions (Koike-Yusa we present methods for the application of the CISPR-Cas9 system et al, 2013; Shalem et al, 2014; Wang et al, 2014). However, in large-scale, image-based, gene perturbation experiments. We pooled screening precludes multivariate profiling of single-cell show that CRISPR-Cas9-mediated gene perturbation can be phenotypes. This can be partially overcome by combining pooled achieved in human tissue culture cells in a timeframe that is screening with single-cell RNA-seq, but this does not easily scale compatible with image-based phenotyping. We developed a pipe- to the profiling of thousands of single cells from thousands of line to construct a large-scale arrayed library of 2,281 sequence- perturbations, and is limited to features that can be read from verified CRISPR-Cas9 targeting plasmids and profiled this library RNA transcript profiles (Adamson et al, 2016; Dixit et al, 2016; for genes affecting cellular morphology and the subcellular local- Jaitin et al, 2016; Datlinger et al, 2017). Moreover, sequencing- ization of components of the nuclear pore complex (NPC). -

Comparative Proteomics Analysis of Human Liver Microsomes and S9
DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Comparative Proteomics Analysis of Human Liver Microsomes and S9 Fractions Xinwen Wang, Bing He, Jian Shi, Qian Li, and Hao-Jie Zhu Department of Clinical Pharmacy, University of Michigan, Ann Arbor, Michigan (X.W., B.H., J.S., H.-J.Z.); and School of Life Science and Technology, China Pharmaceutical University, Nanjing, Jiangsu, 210009 (Q.L.) Downloaded from dmd.aspetjournals.org at ASPET Journals on October 2, 2021 1 DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Running title: Comparative Proteomics of Human Liver Microsomes and S9 Corresponding author: Hao-Jie Zhu Ph.D. Department of Clinical Pharmacy University of Michigan College of Pharmacy 428 Church Street, Room 4565 Downloaded from Ann Arbor, MI 48109-1065 Tel: 734-763-8449, E-mail: [email protected] dmd.aspetjournals.org Number of words in Abstract: 250 at ASPET Journals on October 2, 2021 Number of words in Introduction: 776 Number of words in Discussion: 2304 2 DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Non-standard ABBreviations: DMEs, drug metabolism enzymes; HLM, human liver microsomes; HLS9, -

Functional Gene Clusters in Global Pathogenesis of Clear Cell Carcinoma of the Ovary Discovered by Integrated Analysis of Transcriptomes
International Journal of Environmental Research and Public Health Article Functional Gene Clusters in Global Pathogenesis of Clear Cell Carcinoma of the Ovary Discovered by Integrated Analysis of Transcriptomes Yueh-Han Hsu 1,2, Peng-Hui Wang 1,2,3,4,5 and Chia-Ming Chang 1,2,* 1 Department of Obstetrics and Gynecology, Taipei Veterans General Hospital, Taipei 112, Taiwan; [email protected] (Y.-H.H.); [email protected] (P.-H.W.) 2 School of Medicine, National Yang-Ming University, Taipei 112, Taiwan 3 Institute of Clinical Medicine, National Yang-Ming University, Taipei 112, Taiwan 4 Department of Medical Research, China Medical University Hospital, Taichung 440, Taiwan 5 Female Cancer Foundation, Taipei 104, Taiwan * Correspondence: [email protected]; Tel.: +886-2-2875-7826; Fax: +886-2-5570-2788 Received: 27 April 2020; Accepted: 31 May 2020; Published: 2 June 2020 Abstract: Clear cell carcinoma of the ovary (ovarian clear cell carcinoma (OCCC)) is one epithelial ovarian carcinoma that is known to have a poor prognosis and a tendency for being refractory to treatment due to unclear pathogenesis. Published investigations of OCCC have mainly focused only on individual genes and lack of systematic integrated research to analyze the pathogenesis of OCCC in a genome-wide perspective. Thus, we conducted an integrated analysis using transcriptome datasets from a public domain database to determine genes that may be implicated in the pathogenesis involved in OCCC carcinogenesis. We used the data obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) DataSets. We found six interactive functional gene clusters in the pathogenesis network of OCCC, including ribosomal protein, eukaryotic translation initiation factors, lactate, prostaglandin, proteasome, and insulin-like growth factor. -

Comparative Transcriptomics Identifies Potential Stemness-Related Markers for Mesenchymal Stromal/Stem Cells
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.25.445659; this version posted May 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Comparative Transcriptomics Identifies Potential Stemness-Related Markers for Mesenchymal Stromal/Stem Cells Authors Myret Ghabriel 1, Ahmed El Hosseiny 1, 2, Ahmed Moustafa*1, 2 and Asma Amleh*1, 2 Affiliations 1Biotechnology Program, American University in Cairo, New Cairo 11835, Egypt 2Department of Biology, American University in Cairo, New Cairo 11835, Egypt *Corresponding authors: Ahmed Moustafa [email protected] Asma Amleh [email protected]. Abstract Mesenchymal stromal/stem cells (MSCs) are multipotent cells residing in multiple tissues with the capacity for self-renewal and differentiation into various cell types. These properties make them promising candidates for regenerative therapies. MSC identification is critical in yielding pure populations for successful therapeutic applications; however, the criteria for MSC identification proposed by the International Society for Cellular Therapy (ISCT) is inconsistent across different tissue sources. In this study, we aimed to identify potential markers to be used together with the ISCT’s criteria to provide a more accurate means of MSC identification. Thus, we carried out a comparative analysis of the expression of human and mouse MSCs derived from multiple tissues to identify the common differentially expressed genes. We show that six members of the proteasome degradation system are similarly expressed across MSCs derived from bone marrow, adipose tissue, amnion, and umbilical cord. -

Genome-Wide Transcript and Protein Analysis Reveals Distinct Features of Aging in the Mouse Heart
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.28.272260; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide transcript and protein analysis reveals distinct features of aging in the mouse heart Isabela Gerdes Gyuricza1, Joel M. Chick2, Gregory R. Keele1, Andrew G. Deighan1, Steven C. Munger1, Ron Korstanje1, Steven P. Gygi3, Gary A. Churchill1 1The Jackson Laboratory, Bar Harbor, Maine 04609 USA; 2Vividion Therapeutics, San Diego, California 92121, USA; 3Harvard Medical School, Boston, Massachusetts 02115, USA Corresponding author: [email protected] Key words for online indexing: Heart Aging Transcriptomics Proteomics eQTL pQTL Stoichiometry ABSTRACT Investigation of the molecular mechanisms of aging in the human heart is challenging due to confounding factors, such as diet and medications, as well limited access to tissues. The laboratory mouse provides an ideal model to study aging in healthy individuals in a controlled environment. However, previous mouse studies have examined only a narrow range of the genetic variation that shapes individual differences during aging. Here, we analyzed transcriptome and proteome data from hearts of genetically diverse mice at ages 6, 12 and 18 months to characterize molecular changes that occur in the aging heart. Transcripts and proteins reveal distinct biological processes that are altered through the course of natural aging. Transcriptome analysis reveals a scenario of cardiac hypertrophy, fibrosis, and reemergence of fetal gene expression patterns. -
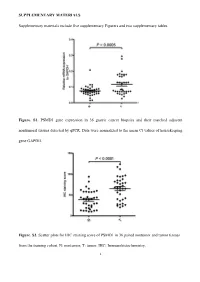
Supplementary Figures and Tables
SUPPLEMENTARY MATERIALS Supplementary materials include five supplementary Figurers and two supplementary tables. Figure. S1. PSMD1 gene expression in 36 gastric cancer biopsies and their matched adjacent nontumoral tissues detected by qPCR. Data were normalized to the mean Ct values of housekeeping gene GAPDH. Figure. S2. Scatter plots for IHC staining score of PSMD1 in 36 paired nontumor and tumor tissues from the training cohort. N: nontumor; T: tumor. IHC: Immunohistochemistry. 1 Figure. S3. Kaplan-Meier survival curve analysis of DFS for all 411 patients with gastric cancer according to the PSMD1 expression stratified by clinicopathological risk factors. P-values were calculated by the log-rank test. 2 Figure. S4. Kaplan-Meier survival curve analysis of OS for all 411 patients with gastric cancer according to the PSMD1 expression stratified by clinicopathological risk factors. P-values were calculated by the log-rank test. 3 Figure. S5. Extended model for prediction of DFS and OS in patients with GC based on PSMD1 expression. ROC analyses of sensitivity and specificity for prediction of a, b DFS and c, d OS by the combined model (TNM stage and PSMD1 expression combination), TNM stage model, andPSMD1 expression model. 4 Figure. S6. X-tile plots of the DFS and OS nomograms. Color intensity of the plot represents low (dark, black) to high (bright, red, or green) strength of association at each division. Red represents the inverse association between the PSMD1 expression and survival (DFS or OS), whereas green represents direct association. a DFS nomogram, b OS nomogram. 5 Table S1. Clinical characteristics of the 36 patients with gastric cancer. -

A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection
CLINICAL RESEARCH www.jasn.org A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection Weijia Zhang,1 Zhengzi Yi,1 Karen L. Keung,2 Huimin Shang,3 Chengguo Wei,1 Paolo Cravedi,1 Zeguo Sun,1 Caixia Xi,1 Christopher Woytovich,1 Samira Farouk,1 Weiqing Huang,1 Khadija Banu,1 Lorenzo Gallon,4 Ciara N. Magee,5 Nader Najafian,5 Milagros Samaniego,6 Arjang Djamali ,7 Stephen I. Alexander,2 Ivy A. Rosales,8 Rex Neal Smith,8 Jenny Xiang,3 Evelyne Lerut,9 Dirk Kuypers,10,11 Maarten Naesens ,10,11 Philip J. O’Connell,2 Robert Colvin,8 Madhav C. Menon,1 and Barbara Murphy1 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background In kidney transplant recipients, surveillance biopsies can reveal, despite stable graft function, histologic features of acute rejection and borderline changes that are associated with undesirable graft outcomes. Noninvasive biomarkers of subclinical acute rejection are needed to avoid the risks and costs associated with repeated biopsies. Methods We examined subclinical histologic and functional changes in kidney transplant recipients from the prospective Genomics of Chronic Allograft Rejection (GoCAR) study who underwent surveillance biopsies over 2 years, identifying those with subclinical or borderline acute cellular rejection (ACR) at 3 months (ACR-3) post-transplant. We performed RNA sequencing on whole blood collected from 88 indi- viduals at the time of 3-month surveillance biopsy to identify transcripts associated with ACR-3, developed a novel sequencing-based targeted expression assay, and validated this gene signature in an independent cohort. -

82318457.Pdf
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Effects of Etanercept Are Distinct from Infliximab in Modulating Proinflammatory Genes in Activated Human Leukocytes Asifa S. Haider1, Irma R. Cardinale1, Julia A. Whynot1 and James G. Krueger1 Proinflammatory diseases like rheumatoid arthritis, Crohn’s disease, and psoriasis have been treated by the tumor necrosis factor (TNF) antagonists infliximab and etanercept with different degrees of success. Although these agents are widely used in humans, little is known about their mechanisms of action or why etanercept and infliximab have differences in clinical activity. In this study, we define leukocyte genes that are suppressed by etanercept within 24 hours of exposure. Compared to previous work with infliximab, fewer immune-related genes are suppressed by etanercept. Importantly, the range of genes suppressed by these alternative TNF inhibitors is only partially overlapping, suggesting each has unique immune modulating effects. In sharp contrast to etanercept, infliximab strongly suppresses genes associated with ‘‘Type 1’’ immune responses (IFN-g and the IL-12-receptor beta 2 subunit), providing a clear mechanism for clinically relevant immune suppression. Journal of Investigative Dermatology Symposium Proceedings (2007) 12, 9–15. doi:10.1038/sj.jidsymp.5650032 INTRODUCTION range of cytokine-regulated gene products is induced. By Two biologic tumor necrosis factor (TNF) inhibitors, etaner- blocking ‘‘inductive’’ effects of secreted cytokines in these cept and infliximab, are effective in treating psoriasis and cultures with monoclonal antibodies, we have been able to psoriatic arthritis (Gottlieb, 2005), but differences exist in the define genes that are selectively regulated by IFN-g or TNF in rapidity of therapeutic improvement, the overall fraction of leukocytes. -

Proteasome Biology: Chemistry and Bioengineering Insights
polymers Review Proteasome Biology: Chemistry and Bioengineering Insights Lucia Raˇcková * and Erika Csekes Centre of Experimental Medicine, Institute of Experimental Pharmacology and Toxicology, Slovak Academy of Sciences, Dúbravská cesta 9, 841 04 Bratislava, Slovakia; [email protected] * Correspondence: [email protected] or [email protected] Received: 28 September 2020; Accepted: 23 November 2020; Published: 4 December 2020 Abstract: Proteasomal degradation provides the crucial machinery for maintaining cellular proteostasis. The biological origins of modulation or impairment of the function of proteasomal complexes may include changes in gene expression of their subunits, ubiquitin mutation, or indirect mechanisms arising from the overall impairment of proteostasis. However, changes in the physico-chemical characteristics of the cellular environment might also meaningfully contribute to altered performance. This review summarizes the effects of physicochemical factors in the cell, such as pH, temperature fluctuations, and reactions with the products of oxidative metabolism, on the function of the proteasome. Furthermore, evidence of the direct interaction of proteasomal complexes with protein aggregates is compared against the knowledge obtained from immobilization biotechnologies. In this regard, factors such as the structures of the natural polymeric scaffolds in the cells, their content of reactive groups or the sequestration of metal ions, and processes at the interface, are discussed here with regard to their -

Genomic and Proteomic Analysis Reveals a Threshold Level of MYC Required for Tumor Maintenance
Research Article Genomic and Proteomic Analysis Reveals a Threshold Level of MYC Required for Tumor Maintenance Catherine M. Shachaf,1,2,3 Andrew J. Gentles,4 Sailaja Elchuri,1 Debashis Sahoo,5 Yoav Soen,6 Orr Sharpe,7 Omar D. Perez,2,3 Maria Chang,1 Dennis Mitchel,2,3 William H. Robinson,7 David Dill,5 Garry P. Nolan,2,3 Sylvia K. Plevritis,4 and Dean W. Felsher1 1Division of Medical Oncology, Departments of Medicine and Pathology, 2Department of Microbiology and Immunology, 3The Baxter Laboratory for Genetic Pharmacology, 4Department of Radiology, 5Department of Computer Science, 6Department of Biochemistry, Howard Hughes Medical Institute, and 7Division of Immunology and Rheumatology, Department of Medicine, Stanford University School of Medicine, Stanford University, Stanford, California Abstract regression in different model systems (7–13). The consequences of MYC overexpression has been implicated in the pathogenesis MYC inactivation in different tumors have shared certain common of most types of human cancers. MYC is likely to contribute to features most notably that on MYC inactivation tumors variously tumorigenesisbyitseffectsonglobalgeneexpression. undergo proliferative arrest, differentiation, and apoptosis. Previously, we have shown that the loss of MYC overexpression MYC has been generally presumed to induce tumorigenesis is sufficient to reverse tumorigenesis. Here, we show that there through its effects as a transcription factor. We hypothesized that is a precise threshold level of MYC expression required for there may be a threshold level of MYC expression required to maintaining the tumor phenotype, whereupon there is a switch maintain a tumor phenotype. At this threshold, there would be from a gene expression program of proliferation to a state of specific changes in gene and protein expression that would define the ability of MYC to function as an oncogene. -

The Kinesin Spindle Protein Inhibitor Filanesib Enhances the Activity of Pomalidomide and Dexamethasone in Multiple Myeloma
Plasma Cell Disorders SUPPLEMENTARY APPENDIX The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma Susana Hernández-García, 1 Laura San-Segundo, 1 Lorena González-Méndez, 1 Luis A. Corchete, 1 Irena Misiewicz- Krzeminska, 1,2 Montserrat Martín-Sánchez, 1 Ana-Alicia López-Iglesias, 1 Esperanza Macarena Algarín, 1 Pedro Mogollón, 1 Andrea Díaz-Tejedor, 1 Teresa Paíno, 1 Brian Tunquist, 3 María-Victoria Mateos, 1 Norma C Gutiérrez, 1 Elena Díaz- Rodriguez, 1 Mercedes Garayoa 1* and Enrique M Ocio 1* 1Centro Investigación del Cáncer-IBMCC (CSIC-USAL) and Hospital Universitario-IBSAL, Salamanca, Spain; 2National Medicines Insti - tute, Warsaw, Poland and 3Array BioPharma, Boulder, Colorado, USA *MG and EMO contributed equally to this work ©2017 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2017.168666 Received: March 13, 2017. Accepted: August 29, 2017. Pre-published: August 31, 2017. Correspondence: [email protected] MATERIAL AND METHODS Reagents and drugs. Filanesib (F) was provided by Array BioPharma Inc. (Boulder, CO, USA). Thalidomide (T), lenalidomide (L) and pomalidomide (P) were purchased from Selleckchem (Houston, TX, USA), dexamethasone (D) from Sigma-Aldrich (St Louis, MO, USA) and bortezomib from LC Laboratories (Woburn, MA, USA). Generic chemicals were acquired from Sigma Chemical Co., Roche Biochemicals (Mannheim, Germany), Merck & Co., Inc. (Darmstadt, Germany). MM cell lines, patient samples and cultures. Origin, authentication and in vitro growth conditions of human MM cell lines have already been characterized (17, 18). The study of drug activity in the presence of IL-6, IGF-1 or in co-culture with primary bone marrow mesenchymal stromal cells (BMSCs) or the human mesenchymal stromal cell line (hMSC–TERT) was performed as described previously (19, 20).