Proteasome 26S Subunit, Non-Atpases 1 (PSMD1) and 3 (PSMD3), Play an Oncogenic Role in Chronic Myeloid Leukemia by Stabilizing Nuclear Factor-Kappa B
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Essentiality Landscape and Druggable Oncogenic Dependencies in Herpesviral Primary Effusion Lymphoma
ARTICLE DOI: 10.1038/s41467-018-05506-9 OPEN Gene essentiality landscape and druggable oncogenic dependencies in herpesviral primary effusion lymphoma Mark Manzano1, Ajinkya Patil1, Alexander Waldrop2, Sandeep S. Dave2, Amir Behdad3 & Eva Gottwein1 Primary effusion lymphoma (PEL) is caused by Kaposi’s sarcoma-associated herpesvirus. Our understanding of PEL is poor and therefore treatment strategies are lacking. To address this 1234567890():,; need, we conducted genome-wide CRISPR/Cas9 knockout screens in eight PEL cell lines. Integration with data from unrelated cancers identifies 210 genes as PEL-specific oncogenic dependencies. Genetic requirements of PEL cell lines are largely independent of Epstein-Barr virus co-infection. Genes of the NF-κB pathway are individually non-essential. Instead, we demonstrate requirements for IRF4 and MDM2. PEL cell lines depend on cellular cyclin D2 and c-FLIP despite expression of viral homologs. Moreover, PEL cell lines are addicted to high levels of MCL1 expression, which are also evident in PEL tumors. Strong dependencies on cyclin D2 and MCL1 render PEL cell lines highly sensitive to palbociclib and S63845. In summary, this work comprehensively identifies genetic dependencies in PEL cell lines and identifies novel strategies for therapeutic intervention. 1 Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. 2 Duke Cancer Institute and Center for Genomic and Computational Biology, Duke University, Durham, NC 27708, USA. 3 Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. Correspondence and requests for materials should be addressed to E.G. (email: [email protected]) NATURE COMMUNICATIONS | (2018) 9:3263 | DOI: 10.1038/s41467-018-05506-9 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-05506-9 he human oncogenic γ-herpesvirus Kaposi’s sarcoma- (IRF4), a critical oncogene in multiple myeloma33. -

Large-Scale Image-Based Profiling of Single-Cell Phenotypes in Arrayed CRISPR-Cas9 Gene Perturbation Screens
Published online: January 23, 2018 Method Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR-Cas9 gene perturbation screens Reinoud de Groot1 , Joel Lüthi1,2 , Helen Lindsay1 , René Holtackers1 & Lucas Pelkmans1,* Abstract this reason, CRISPR-Cas9 has been used in large-scale functional genomic screens (Shalem et al, 2015). Most screens performed to High-content imaging using automated microscopy and computer date employ a pooled screening strategy, which can identify genes vision allows multivariate profiling of single-cell phenotypes. Here, that cause differential growth in screening conditions (Koike-Yusa we present methods for the application of the CISPR-Cas9 system et al, 2013; Shalem et al, 2014; Wang et al, 2014). However, in large-scale, image-based, gene perturbation experiments. We pooled screening precludes multivariate profiling of single-cell show that CRISPR-Cas9-mediated gene perturbation can be phenotypes. This can be partially overcome by combining pooled achieved in human tissue culture cells in a timeframe that is screening with single-cell RNA-seq, but this does not easily scale compatible with image-based phenotyping. We developed a pipe- to the profiling of thousands of single cells from thousands of line to construct a large-scale arrayed library of 2,281 sequence- perturbations, and is limited to features that can be read from verified CRISPR-Cas9 targeting plasmids and profiled this library RNA transcript profiles (Adamson et al, 2016; Dixit et al, 2016; for genes affecting cellular morphology and the subcellular local- Jaitin et al, 2016; Datlinger et al, 2017). Moreover, sequencing- ization of components of the nuclear pore complex (NPC). -

Comparative Proteomics Analysis of Human Liver Microsomes and S9
DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Comparative Proteomics Analysis of Human Liver Microsomes and S9 Fractions Xinwen Wang, Bing He, Jian Shi, Qian Li, and Hao-Jie Zhu Department of Clinical Pharmacy, University of Michigan, Ann Arbor, Michigan (X.W., B.H., J.S., H.-J.Z.); and School of Life Science and Technology, China Pharmaceutical University, Nanjing, Jiangsu, 210009 (Q.L.) Downloaded from dmd.aspetjournals.org at ASPET Journals on October 2, 2021 1 DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Running title: Comparative Proteomics of Human Liver Microsomes and S9 Corresponding author: Hao-Jie Zhu Ph.D. Department of Clinical Pharmacy University of Michigan College of Pharmacy 428 Church Street, Room 4565 Downloaded from Ann Arbor, MI 48109-1065 Tel: 734-763-8449, E-mail: [email protected] dmd.aspetjournals.org Number of words in Abstract: 250 at ASPET Journals on October 2, 2021 Number of words in Introduction: 776 Number of words in Discussion: 2304 2 DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Non-standard ABBreviations: DMEs, drug metabolism enzymes; HLM, human liver microsomes; HLS9, -

Produktinformation
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic PSMD10 Antibody, Biotin conjugated Product Code CSB-PA018899LD01HU Abbreviation 26S proteasome non-ATPase regulatory subunit 10 Storage Upon receipt, store at -20°C or -80°C. Avoid repeated freeze. Uniprot No. O75832 Immunogen Recombinant Human 26S proteasome non-ATPase regulatory subunit 10 protein (1-226AA) Raised In Rabbit Species Reactivity Human Tested Applications ELISA Relevance Acts as a chaperone during the assembly of the 26S proteasome, specifically of the PA700/19S regulatory complex (RC). In the initial step of the base subcomplex assembly is part of an intermediate PSMD10:PSMC4:PSMC5:PAAF1 module which probably assembles with a PSMD5:PSMC2:PSMC1:PSMD2 module. Independently of the proteasome, regulates EGF-induced AKT activation through inhibition of the RHOA/ROCK/PTEN pahway, leading to prolonged AKT activation. Plays an important role in RAS-induced tumorigenesis. Acts as an proto-oncoprotein by being involved in negative regulation of tumor suppressors RB1 and p53/TP53. Overexpression is leading to phosphorylation of RB1 and proteasomal degradation of RB1. Regulates CDK4-mediated phosphorylation of RB1 by competing with CDKN2A for binding with CDK4. Facilitates binding of MDM2 to p53/TP53 and the mono- and polyubiquitination of p53/TP53 by MDM2 suggesting a function in targeting the TP53:MDM2 complex to the 26S proteasome. -

Cancer Vulnerabilities Unveiled by Genomic Loss
Cancer Vulnerabilities Unveiled by Genomic Loss The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Nijhawan, Deepak, Travis I. Zack, Yin Ren, Matthew R. Strickland, Rebecca Lamothe, Steven E. Schumacher, Aviad Tsherniak, et al. “Cancer Vulnerabilities Unveiled by Genomic Loss.” Cell 150, no. 4 (August 2012): 842–854. © 2012 Elsevier Inc. As Published http://dx.doi.org/10.1016/j.cell.2012.07.023 Publisher Elsevier B.V. Version Final published version Citable link http://hdl.handle.net/1721.1/91899 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Cancer Vulnerabilities Unveiled by Genomic Loss Deepak Nijhawan,1,2,7,9,10 Travis I. Zack,1,2,3,9 Yin Ren,5 Matthew R. Strickland,1 Rebecca Lamothe,1 Steven E. Schumacher,1,2 Aviad Tsherniak,2 Henrike C. Besche,4 Joseph Rosenbluh,1,2,7 Shyemaa Shehata,1 Glenn S. Cowley,2 Barbara A. Weir,2 Alfred L. Goldberg,4 Jill P. Mesirov,2 David E. Root,2 Sangeeta N. Bhatia,2,5,6,7,8 Rameen Beroukhim,1,2,7,* and William C. Hahn1,2,7,* 1Departments of Cancer Biology and Medical Oncology, Dana Farber Cancer Institute, Boston, MA 02215, USA 2Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA 3Biophysics Program, Harvard University, Boston, MA 02115, USA 4Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115, USA 5Harvard-MIT Division of Health Sciences and Technology 6David H. -

PSMD10 Antibody (Center) Purified Rabbit Polyclonal Antibody (Pab) Catalog # AW5126
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 PSMD10 Antibody (Center) Purified Rabbit Polyclonal Antibody (Pab) Catalog # AW5126 Specification PSMD10 Antibody (Center) - Product Information Application IF, WB, IHC-P,E Primary Accession O75832 Other Accession Q9Z2X2 Reactivity Human Predicted Mouse Host Rabbit Clonality Polyclonal Calculated MW H=24,16;M=25;Ra t=25 KDa Isotype Rabbit Ig Antigen Source HUMAN PSMD10 Antibody (Center) - Additional Information Gene ID 5716 Fluorescent image of Hela cells stained with PSMD10 Antibody (Center)(Cat#AW5126). Antigen Region AW5126 was diluted at 1:25 dilution. An 43-76 Alexa Fluor 488-conjugated goat anti-rabbit lgG at 1:400 dilution was used as the Other Names secondary antibody (green). Cytoplasmic 26S proteasome non-ATPase regulatory subunit 10, 26S proteasome regulatory actin was counterstained with Alexa Fluor® subunit p28, Gankyrin, p28(GANK), PSMD10 555 conjugated with Phalloidin (red). Dilution IF~~1:25 WB~~ 1:1000 IHC-P~~1:25 Target/Specificity This PSMD10 antibody is generated from a rabbit immunized with a KLH conjugated synthetic peptide between 43-76 amino acids from the Central region of human PSMD10. Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein A column, followed by peptide affinity purification. Western blot analysis of lysates from MCF-7, PC-3, K562 cell line (from left to right), using Storage PSMD10 Antibody (Center)(Cat. #AW5126). Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Maintain refrigerated at 2-8°C for up to 2 AW5126 was diluted at 1:1000 at each lane. -

Functional Gene Clusters in Global Pathogenesis of Clear Cell Carcinoma of the Ovary Discovered by Integrated Analysis of Transcriptomes
International Journal of Environmental Research and Public Health Article Functional Gene Clusters in Global Pathogenesis of Clear Cell Carcinoma of the Ovary Discovered by Integrated Analysis of Transcriptomes Yueh-Han Hsu 1,2, Peng-Hui Wang 1,2,3,4,5 and Chia-Ming Chang 1,2,* 1 Department of Obstetrics and Gynecology, Taipei Veterans General Hospital, Taipei 112, Taiwan; [email protected] (Y.-H.H.); [email protected] (P.-H.W.) 2 School of Medicine, National Yang-Ming University, Taipei 112, Taiwan 3 Institute of Clinical Medicine, National Yang-Ming University, Taipei 112, Taiwan 4 Department of Medical Research, China Medical University Hospital, Taichung 440, Taiwan 5 Female Cancer Foundation, Taipei 104, Taiwan * Correspondence: [email protected]; Tel.: +886-2-2875-7826; Fax: +886-2-5570-2788 Received: 27 April 2020; Accepted: 31 May 2020; Published: 2 June 2020 Abstract: Clear cell carcinoma of the ovary (ovarian clear cell carcinoma (OCCC)) is one epithelial ovarian carcinoma that is known to have a poor prognosis and a tendency for being refractory to treatment due to unclear pathogenesis. Published investigations of OCCC have mainly focused only on individual genes and lack of systematic integrated research to analyze the pathogenesis of OCCC in a genome-wide perspective. Thus, we conducted an integrated analysis using transcriptome datasets from a public domain database to determine genes that may be implicated in the pathogenesis involved in OCCC carcinogenesis. We used the data obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) DataSets. We found six interactive functional gene clusters in the pathogenesis network of OCCC, including ribosomal protein, eukaryotic translation initiation factors, lactate, prostaglandin, proteasome, and insulin-like growth factor. -

Correction1 4784..4785
Correction Correction: PCI-24781 Induces Caspase and Reactive Oxygen Species-Dependent Apoptosis In the article on PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis published in the May 15, 2009 issue of Clinical Cancer Research, there was an error in Table 1. Down-regulated genes were incorrectly labeled as up-regulated genes. The correct table appears here. Bhalla S, Balasubramanian S, David K, et al. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-nB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res 2009;15:3354–65. Table 1. Selected genes from expression analysis following 24-h treatment with PCI-24781, bortezomib, or the combination (in Ramos cells) Accn # Down-regulated genes 0.25 Mmol/L PCI/3 nmol/L Bor Name PCI-24781 Bortezomib Combination* Cell cycle-related NM_000075 Cyclin-dependent kinase 4 (CDK4) 0.49 0.83 0.37 NM_001237 Cyclin A2 (CCNA2) 0.43 0.87 0.37 NM_001950 E2F transcription factor 4, p107/p130-binding (E2F4) 0.48 0.79 0.40 NM_001951 E2F transcription factor 5, p130-binding (E2F5) 0.46 0.98 0.43 NM_003903 CDC16 cell division cycle 16 homolog (S cerevisiae) (CDC16) 0.61 0.78 0.43 NM_031966 Cyclin B1 (CCNB1) 0.55 0.90 0.43 NM_001760 Cyclin D3 (CCND3) 0.48 1.02 0.46 NM_001255 CDC20 cell division cycle 20 homolog (S cerevisiae; CDC20) 0.61 0.82 0.46 NM_001262 Cyclin-dependent kinase inhibitor 2C (p18, inhibits CDK4; CDKN2C) 0.61 1.15 0.56 NM_001238 Cyclin E1 (CCNE1) 0.56 1.05 0.60 NM_001239 Cyclin H (CCNH) 0.74 0.90 0.64 NM_004701 -

Comparative Transcriptomics Identifies Potential Stemness-Related Markers for Mesenchymal Stromal/Stem Cells
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.25.445659; this version posted May 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Comparative Transcriptomics Identifies Potential Stemness-Related Markers for Mesenchymal Stromal/Stem Cells Authors Myret Ghabriel 1, Ahmed El Hosseiny 1, 2, Ahmed Moustafa*1, 2 and Asma Amleh*1, 2 Affiliations 1Biotechnology Program, American University in Cairo, New Cairo 11835, Egypt 2Department of Biology, American University in Cairo, New Cairo 11835, Egypt *Corresponding authors: Ahmed Moustafa [email protected] Asma Amleh [email protected]. Abstract Mesenchymal stromal/stem cells (MSCs) are multipotent cells residing in multiple tissues with the capacity for self-renewal and differentiation into various cell types. These properties make them promising candidates for regenerative therapies. MSC identification is critical in yielding pure populations for successful therapeutic applications; however, the criteria for MSC identification proposed by the International Society for Cellular Therapy (ISCT) is inconsistent across different tissue sources. In this study, we aimed to identify potential markers to be used together with the ISCT’s criteria to provide a more accurate means of MSC identification. Thus, we carried out a comparative analysis of the expression of human and mouse MSCs derived from multiple tissues to identify the common differentially expressed genes. We show that six members of the proteasome degradation system are similarly expressed across MSCs derived from bone marrow, adipose tissue, amnion, and umbilical cord. -

Genome-Wide Transcript and Protein Analysis Reveals Distinct Features of Aging in the Mouse Heart
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.28.272260; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide transcript and protein analysis reveals distinct features of aging in the mouse heart Isabela Gerdes Gyuricza1, Joel M. Chick2, Gregory R. Keele1, Andrew G. Deighan1, Steven C. Munger1, Ron Korstanje1, Steven P. Gygi3, Gary A. Churchill1 1The Jackson Laboratory, Bar Harbor, Maine 04609 USA; 2Vividion Therapeutics, San Diego, California 92121, USA; 3Harvard Medical School, Boston, Massachusetts 02115, USA Corresponding author: [email protected] Key words for online indexing: Heart Aging Transcriptomics Proteomics eQTL pQTL Stoichiometry ABSTRACT Investigation of the molecular mechanisms of aging in the human heart is challenging due to confounding factors, such as diet and medications, as well limited access to tissues. The laboratory mouse provides an ideal model to study aging in healthy individuals in a controlled environment. However, previous mouse studies have examined only a narrow range of the genetic variation that shapes individual differences during aging. Here, we analyzed transcriptome and proteome data from hearts of genetically diverse mice at ages 6, 12 and 18 months to characterize molecular changes that occur in the aging heart. Transcripts and proteins reveal distinct biological processes that are altered through the course of natural aging. Transcriptome analysis reveals a scenario of cardiac hypertrophy, fibrosis, and reemergence of fetal gene expression patterns. -
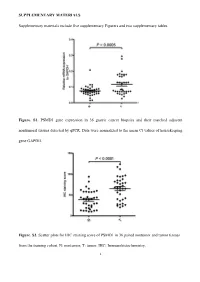
Supplementary Figures and Tables
SUPPLEMENTARY MATERIALS Supplementary materials include five supplementary Figurers and two supplementary tables. Figure. S1. PSMD1 gene expression in 36 gastric cancer biopsies and their matched adjacent nontumoral tissues detected by qPCR. Data were normalized to the mean Ct values of housekeeping gene GAPDH. Figure. S2. Scatter plots for IHC staining score of PSMD1 in 36 paired nontumor and tumor tissues from the training cohort. N: nontumor; T: tumor. IHC: Immunohistochemistry. 1 Figure. S3. Kaplan-Meier survival curve analysis of DFS for all 411 patients with gastric cancer according to the PSMD1 expression stratified by clinicopathological risk factors. P-values were calculated by the log-rank test. 2 Figure. S4. Kaplan-Meier survival curve analysis of OS for all 411 patients with gastric cancer according to the PSMD1 expression stratified by clinicopathological risk factors. P-values were calculated by the log-rank test. 3 Figure. S5. Extended model for prediction of DFS and OS in patients with GC based on PSMD1 expression. ROC analyses of sensitivity and specificity for prediction of a, b DFS and c, d OS by the combined model (TNM stage and PSMD1 expression combination), TNM stage model, andPSMD1 expression model. 4 Figure. S6. X-tile plots of the DFS and OS nomograms. Color intensity of the plot represents low (dark, black) to high (bright, red, or green) strength of association at each division. Red represents the inverse association between the PSMD1 expression and survival (DFS or OS), whereas green represents direct association. a DFS nomogram, b OS nomogram. 5 Table S1. Clinical characteristics of the 36 patients with gastric cancer. -

A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection
CLINICAL RESEARCH www.jasn.org A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection Weijia Zhang,1 Zhengzi Yi,1 Karen L. Keung,2 Huimin Shang,3 Chengguo Wei,1 Paolo Cravedi,1 Zeguo Sun,1 Caixia Xi,1 Christopher Woytovich,1 Samira Farouk,1 Weiqing Huang,1 Khadija Banu,1 Lorenzo Gallon,4 Ciara N. Magee,5 Nader Najafian,5 Milagros Samaniego,6 Arjang Djamali ,7 Stephen I. Alexander,2 Ivy A. Rosales,8 Rex Neal Smith,8 Jenny Xiang,3 Evelyne Lerut,9 Dirk Kuypers,10,11 Maarten Naesens ,10,11 Philip J. O’Connell,2 Robert Colvin,8 Madhav C. Menon,1 and Barbara Murphy1 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background In kidney transplant recipients, surveillance biopsies can reveal, despite stable graft function, histologic features of acute rejection and borderline changes that are associated with undesirable graft outcomes. Noninvasive biomarkers of subclinical acute rejection are needed to avoid the risks and costs associated with repeated biopsies. Methods We examined subclinical histologic and functional changes in kidney transplant recipients from the prospective Genomics of Chronic Allograft Rejection (GoCAR) study who underwent surveillance biopsies over 2 years, identifying those with subclinical or borderline acute cellular rejection (ACR) at 3 months (ACR-3) post-transplant. We performed RNA sequencing on whole blood collected from 88 indi- viduals at the time of 3-month surveillance biopsy to identify transcripts associated with ACR-3, developed a novel sequencing-based targeted expression assay, and validated this gene signature in an independent cohort.