Genomic and Proteomic Analysis Reveals a Threshold Level of MYC Required for Tumor Maintenance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Essentiality Landscape and Druggable Oncogenic Dependencies in Herpesviral Primary Effusion Lymphoma
ARTICLE DOI: 10.1038/s41467-018-05506-9 OPEN Gene essentiality landscape and druggable oncogenic dependencies in herpesviral primary effusion lymphoma Mark Manzano1, Ajinkya Patil1, Alexander Waldrop2, Sandeep S. Dave2, Amir Behdad3 & Eva Gottwein1 Primary effusion lymphoma (PEL) is caused by Kaposi’s sarcoma-associated herpesvirus. Our understanding of PEL is poor and therefore treatment strategies are lacking. To address this 1234567890():,; need, we conducted genome-wide CRISPR/Cas9 knockout screens in eight PEL cell lines. Integration with data from unrelated cancers identifies 210 genes as PEL-specific oncogenic dependencies. Genetic requirements of PEL cell lines are largely independent of Epstein-Barr virus co-infection. Genes of the NF-κB pathway are individually non-essential. Instead, we demonstrate requirements for IRF4 and MDM2. PEL cell lines depend on cellular cyclin D2 and c-FLIP despite expression of viral homologs. Moreover, PEL cell lines are addicted to high levels of MCL1 expression, which are also evident in PEL tumors. Strong dependencies on cyclin D2 and MCL1 render PEL cell lines highly sensitive to palbociclib and S63845. In summary, this work comprehensively identifies genetic dependencies in PEL cell lines and identifies novel strategies for therapeutic intervention. 1 Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. 2 Duke Cancer Institute and Center for Genomic and Computational Biology, Duke University, Durham, NC 27708, USA. 3 Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. Correspondence and requests for materials should be addressed to E.G. (email: [email protected]) NATURE COMMUNICATIONS | (2018) 9:3263 | DOI: 10.1038/s41467-018-05506-9 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-05506-9 he human oncogenic γ-herpesvirus Kaposi’s sarcoma- (IRF4), a critical oncogene in multiple myeloma33. -

SUMO-1 Regulates the Conformational Dynamics of Thymine-DNA
Smet-Nocca et al. BMC Biochemistry 2011, 12:4 http://www.biomedcentral.com/1471-2091/12/4 RESEARCHARTICLE Open Access SUMO-1 regulates the conformational dynamics of Thymine-DNA Glycosylase regulatory domain and competes with its DNA binding activity Caroline Smet-Nocca1, Jean-Michel Wieruszeski2, Hélène Léger1,3, Sebastian Eilebrecht1,3, Arndt Benecke1,3* Abstract Background: The human thymine-DNA glycosylase (TDG) plays a dual role in base excision repair of G:U/T mismatches and in transcription. Regulation of TDG activity by SUMO-1 conjugation was shown to act on both functions. Furthermore, TDG can interact with SUMO-1 in a non-covalent manner. Results: Using NMR spectroscopy we have determined distinct conformational changes in TDG upon either covalent sumoylation on lysine 330 or intermolecular SUMO-1 binding through a unique SUMO-binding motif (SBM) localized in the C-terminal region of TDG. The non-covalent SUMO-1 binding induces a conformational change of the TDG amino-terminal regulatory domain (RD). Such conformational dynamics do not exist with covalent SUMO-1 attachment and could potentially play a broader role in the regulation of TDG functions for instance during transcription. Both covalent and non-covalent processes activate TDG G:U repair similarly. Surprisingly, despite a dissociation of the SBM/SUMO-1 complex in presence of a DNA substrate, SUMO-1 preserves its ability to stimulate TDG activity indicating that the non-covalent interactions are not directly involved in the regulation of TDG activity. SUMO-1 instead acts, as demonstrated here, indirectly by competing with the regulatory domain of TDG for DNA binding. -

Large-Scale Image-Based Profiling of Single-Cell Phenotypes in Arrayed CRISPR-Cas9 Gene Perturbation Screens
Published online: January 23, 2018 Method Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR-Cas9 gene perturbation screens Reinoud de Groot1 , Joel Lüthi1,2 , Helen Lindsay1 , René Holtackers1 & Lucas Pelkmans1,* Abstract this reason, CRISPR-Cas9 has been used in large-scale functional genomic screens (Shalem et al, 2015). Most screens performed to High-content imaging using automated microscopy and computer date employ a pooled screening strategy, which can identify genes vision allows multivariate profiling of single-cell phenotypes. Here, that cause differential growth in screening conditions (Koike-Yusa we present methods for the application of the CISPR-Cas9 system et al, 2013; Shalem et al, 2014; Wang et al, 2014). However, in large-scale, image-based, gene perturbation experiments. We pooled screening precludes multivariate profiling of single-cell show that CRISPR-Cas9-mediated gene perturbation can be phenotypes. This can be partially overcome by combining pooled achieved in human tissue culture cells in a timeframe that is screening with single-cell RNA-seq, but this does not easily scale compatible with image-based phenotyping. We developed a pipe- to the profiling of thousands of single cells from thousands of line to construct a large-scale arrayed library of 2,281 sequence- perturbations, and is limited to features that can be read from verified CRISPR-Cas9 targeting plasmids and profiled this library RNA transcript profiles (Adamson et al, 2016; Dixit et al, 2016; for genes affecting cellular morphology and the subcellular local- Jaitin et al, 2016; Datlinger et al, 2017). Moreover, sequencing- ization of components of the nuclear pore complex (NPC). -

Comparative Proteomics Analysis of Human Liver Microsomes and S9
DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Comparative Proteomics Analysis of Human Liver Microsomes and S9 Fractions Xinwen Wang, Bing He, Jian Shi, Qian Li, and Hao-Jie Zhu Department of Clinical Pharmacy, University of Michigan, Ann Arbor, Michigan (X.W., B.H., J.S., H.-J.Z.); and School of Life Science and Technology, China Pharmaceutical University, Nanjing, Jiangsu, 210009 (Q.L.) Downloaded from dmd.aspetjournals.org at ASPET Journals on October 2, 2021 1 DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Running title: Comparative Proteomics of Human Liver Microsomes and S9 Corresponding author: Hao-Jie Zhu Ph.D. Department of Clinical Pharmacy University of Michigan College of Pharmacy 428 Church Street, Room 4565 Downloaded from Ann Arbor, MI 48109-1065 Tel: 734-763-8449, E-mail: [email protected] dmd.aspetjournals.org Number of words in Abstract: 250 at ASPET Journals on October 2, 2021 Number of words in Introduction: 776 Number of words in Discussion: 2304 2 DMD Fast Forward. Published on November 7, 2019 as DOI: 10.1124/dmd.119.089235 This article has not been copyedited and formatted. The final version may differ from this version. DMD # 89235 Non-standard ABBreviations: DMEs, drug metabolism enzymes; HLM, human liver microsomes; HLS9, -

T-Cell Receptor (TCR) Signaling Promotes the Assembly of Ranbp2
RESEARCH ARTICLE T-cell receptor (TCR) signaling promotes the assembly of RanBP2/RanGAP1- SUMO1/Ubc9 nuclear pore subcomplex via PKC--mediated phosphorylation of RanGAP1 Yujiao He1, Zhiguo Yang1†, Chen-si Zhao1†, Zhihui Xiao1†, Yu Gong1, Yun-Yi Li1, Yiqi Chen1, Yunting Du1, Dianying Feng1, Amnon Altman2, Yingqiu Li1* 1MOE Key Laboratory of Gene Function and Regulation, Guangdong Province Key Laboratory of Pharmaceutical Functional Genes, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou, China; 2Center for Cancer Immunotherapy, La Jolla Institute for Immunology, La Jolla, United States Abstract The nuclear pore complex (NPC) is the sole and selective gateway for nuclear transport, and its dysfunction has been associated with many diseases. The metazoan NPC subcomplex RanBP2, which consists of RanBP2 (Nup358), RanGAP1-SUMO1, and Ubc9, regulates the assembly and function of the NPC. The roles of immune signaling in regulation of NPC remain poorly understood. Here, we show that in human and murine T cells, following T-cell receptor (TCR) stimulation, protein kinase C-q (PKC-q) directly phosphorylates RanGAP1 to facilitate RanBP2 subcomplex assembly and nuclear import and, thus, the nuclear translocation of AP-1 transcription *For correspondence: factor. Mechanistically, TCR stimulation induces the translocation of activated PKC-q to the NPC, 504 506 [email protected] where it interacts with and phosphorylates RanGAP1 on Ser and Ser . RanGAP1 phosphorylation increases its binding affinity for Ubc9, thereby promoting sumoylation of RanGAP1 †These authors contributed and, finally, assembly of the RanBP2 subcomplex. Our findings reveal an unexpected role of PKC-q equally to this work as a direct regulator of nuclear import and uncover a phosphorylation-dependent sumoylation of Competing interests: The RanGAP1, delineating a novel link between TCR signaling and assembly of the RanBP2 NPC authors declare that no subcomplex. -

The E3 Ligase PIAS1 Regulates P53 Sumoylation to Control Stress-Induced Apoptosis of Lens
fcell-09-660494 June 14, 2021 Time: 13:5 # 1 ORIGINAL RESEARCH published: 14 June 2021 doi: 10.3389/fcell.2021.660494 The E3 Ligase PIAS1 Regulates p53 Sumoylation to Control Stress-Induced Apoptosis of Lens Edited by: Wolfgang Knabe, Epithelial Cells Through the Universität Münster, Germany Proapoptotic Regulator Bax Reviewed by: Guillaume Bossis, † † † † † Centre National de la Recherche Qian Nie , Huimin Chen , Ming Zou , Ling Wang , Min Hou , Jia-Wen Xiang, Scientifique (CNRS), France Zhongwen Luo, Xiao-Dong Gong, Jia-Ling Fu, Yan Wang, Shu-Yu Zheng, Yuan Xiao, Stefan Müller, Yu-Wen Gan, Qian Gao, Yue-Yue Bai, Jing-Miao Wang, Lan Zhang, Xiang-Cheng Tang, Goethe University Frankfurt, Germany Xuebin Hu, Lili Gong, Yizhi Liu* and David Wan-Cheng Li*‡ Leena Latonen, University of Eastern Finland, Finland State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China *Correspondence: David Wan-Cheng Li Protein sumoylation is one of the most important post-translational modifications [email protected] Yizhi Liu regulating many biological processes (Flotho A & Melchior F. 2013. Ann Rev. Biochem. [email protected] 82:357–85). Our previous studies have shown that sumoylation plays a fundamental †These authors have contributed role in regulating lens differentiation (Yan et al., 2010. PNAS, 107(49):21034-9.; equally to this work Gong et al., 2014. PNAS. 111(15):5574–9). Whether sumoylation is implicated in lens ‡ ORCID: David Wan-Cheng Li pathogenesis remains elusive. Here, we present evidence to show that the protein orcid.org/0000-0002-7398-7630 inhibitor of activated STAT-1 (PIAS1), a E3 ligase for sumoylation, is implicated in regulating stress-induced lens pathogenesis. -

The Role of Sumoylation of DNA Topoisomerase Iiα C-Terminal Domain in the Regulation of Mitotic Kinases In
SUMOylation at the centromere: The role of SUMOylation of DNA topoisomerase IIα C-terminal domain in the regulation of mitotic kinases in cell cycle progression. By Makoto Michael Yoshida Submitted to the graduate degree program in the Department of Molecular Biosciences and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. ________________________________________ Chairperson: Yoshiaki Azuma, Ph.D. ________________________________________ Roberto De Guzman, Ph.D. ________________________________________ Kristi Neufeld, Ph.D. _________________________________________ Berl Oakley, Ph.D. _________________________________________ Blake Peterson, Ph.D. Date Defended: July 12, 2016 The Dissertation Committee for Makoto Michael Yoshida certifies that this is the approved version of the following dissertation: SUMOylation at the centromere: The role of SUMOylation of DNA topoisomerase IIα C-terminal domain in the regulation of mitotic kinases in cell cycle progression. ________________________________________ Chairperson: Yoshiaki Azuma, Ph.D. Date approved: July 12, 2016 ii ABSTRACT In many model systems, SUMOylation is required for proper mitosis; in particular, chromosome segregation during anaphase. It was previously shown that interruption of SUMOylation through the addition of the dominant negative E2 SUMO conjugating enzyme Ubc9 in mitosis causes abnormal chromosome segregation in Xenopus laevis egg extract (XEE) cell-free assays, and DNA topoisomerase IIα (TOP2A) was identified as a substrate for SUMOylation at the mitotic centromeres. TOP2A is SUMOylated at K660 and multiple sites in the C-terminal domain (CTD). We sought to understand the role of TOP2A SUMOylation at the mitotic centromeres by identifying specific binding proteins for SUMOylated TOP2A CTD. Through affinity isolation, we have identified Haspin, a histone H3 threonine 3 (H3T3) kinase, as a SUMOylated TOP2A CTD binding protein. -

Functional Gene Clusters in Global Pathogenesis of Clear Cell Carcinoma of the Ovary Discovered by Integrated Analysis of Transcriptomes
International Journal of Environmental Research and Public Health Article Functional Gene Clusters in Global Pathogenesis of Clear Cell Carcinoma of the Ovary Discovered by Integrated Analysis of Transcriptomes Yueh-Han Hsu 1,2, Peng-Hui Wang 1,2,3,4,5 and Chia-Ming Chang 1,2,* 1 Department of Obstetrics and Gynecology, Taipei Veterans General Hospital, Taipei 112, Taiwan; [email protected] (Y.-H.H.); [email protected] (P.-H.W.) 2 School of Medicine, National Yang-Ming University, Taipei 112, Taiwan 3 Institute of Clinical Medicine, National Yang-Ming University, Taipei 112, Taiwan 4 Department of Medical Research, China Medical University Hospital, Taichung 440, Taiwan 5 Female Cancer Foundation, Taipei 104, Taiwan * Correspondence: [email protected]; Tel.: +886-2-2875-7826; Fax: +886-2-5570-2788 Received: 27 April 2020; Accepted: 31 May 2020; Published: 2 June 2020 Abstract: Clear cell carcinoma of the ovary (ovarian clear cell carcinoma (OCCC)) is one epithelial ovarian carcinoma that is known to have a poor prognosis and a tendency for being refractory to treatment due to unclear pathogenesis. Published investigations of OCCC have mainly focused only on individual genes and lack of systematic integrated research to analyze the pathogenesis of OCCC in a genome-wide perspective. Thus, we conducted an integrated analysis using transcriptome datasets from a public domain database to determine genes that may be implicated in the pathogenesis involved in OCCC carcinogenesis. We used the data obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) DataSets. We found six interactive functional gene clusters in the pathogenesis network of OCCC, including ribosomal protein, eukaryotic translation initiation factors, lactate, prostaglandin, proteasome, and insulin-like growth factor. -

Quantitative SUMO Proteomics Reveals the Modulation of Several
www.nature.com/scientificreports OPEN Quantitative SUMO proteomics reveals the modulation of several PML nuclear body associated Received: 10 October 2017 Accepted: 28 March 2018 proteins and an anti-senescence Published: xx xx xxxx function of UBC9 Francis P. McManus1, Véronique Bourdeau2, Mariana Acevedo2, Stéphane Lopes-Paciencia2, Lian Mignacca2, Frédéric Lamoliatte1,3, John W. Rojas Pino2, Gerardo Ferbeyre2 & Pierre Thibault1,3 Several regulators of SUMOylation have been previously linked to senescence but most targets of this modifcation in senescent cells remain unidentifed. Using a two-step purifcation of a modifed SUMO3, we profled the SUMO proteome of senescent cells in a site-specifc manner. We identifed 25 SUMO sites on 23 proteins that were signifcantly regulated during senescence. Of note, most of these proteins were PML nuclear body (PML-NB) associated, which correlates with the increased number and size of PML-NBs observed in senescent cells. Interestingly, the sole SUMO E2 enzyme, UBC9, was more SUMOylated during senescence on its Lys-49. Functional studies of a UBC9 mutant at Lys-49 showed a decreased association to PML-NBs and the loss of UBC9’s ability to delay senescence. We thus propose both pro- and anti-senescence functions of protein SUMOylation. Many cellular mechanisms of defense have evolved to reduce the onset of tumors and potential cancer develop- ment. One such mechanism is cellular senescence where cells undergo cell cycle arrest in response to various stressors1,2. Multiple triggers for the onset of senescence have been documented. While replicative senescence is primarily caused in response to telomere shortening3,4, senescence can also be triggered early by a number of exogenous factors including DNA damage, elevated levels of reactive oxygen species (ROS), high cytokine signa- ling, and constitutively-active oncogenes (such as H-RAS-G12V)5,6. -

Comparative Transcriptomics Identifies Potential Stemness-Related Markers for Mesenchymal Stromal/Stem Cells
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.25.445659; this version posted May 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Comparative Transcriptomics Identifies Potential Stemness-Related Markers for Mesenchymal Stromal/Stem Cells Authors Myret Ghabriel 1, Ahmed El Hosseiny 1, 2, Ahmed Moustafa*1, 2 and Asma Amleh*1, 2 Affiliations 1Biotechnology Program, American University in Cairo, New Cairo 11835, Egypt 2Department of Biology, American University in Cairo, New Cairo 11835, Egypt *Corresponding authors: Ahmed Moustafa [email protected] Asma Amleh [email protected]. Abstract Mesenchymal stromal/stem cells (MSCs) are multipotent cells residing in multiple tissues with the capacity for self-renewal and differentiation into various cell types. These properties make them promising candidates for regenerative therapies. MSC identification is critical in yielding pure populations for successful therapeutic applications; however, the criteria for MSC identification proposed by the International Society for Cellular Therapy (ISCT) is inconsistent across different tissue sources. In this study, we aimed to identify potential markers to be used together with the ISCT’s criteria to provide a more accurate means of MSC identification. Thus, we carried out a comparative analysis of the expression of human and mouse MSCs derived from multiple tissues to identify the common differentially expressed genes. We show that six members of the proteasome degradation system are similarly expressed across MSCs derived from bone marrow, adipose tissue, amnion, and umbilical cord. -

Genome-Wide Transcript and Protein Analysis Reveals Distinct Features of Aging in the Mouse Heart
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.28.272260; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide transcript and protein analysis reveals distinct features of aging in the mouse heart Isabela Gerdes Gyuricza1, Joel M. Chick2, Gregory R. Keele1, Andrew G. Deighan1, Steven C. Munger1, Ron Korstanje1, Steven P. Gygi3, Gary A. Churchill1 1The Jackson Laboratory, Bar Harbor, Maine 04609 USA; 2Vividion Therapeutics, San Diego, California 92121, USA; 3Harvard Medical School, Boston, Massachusetts 02115, USA Corresponding author: [email protected] Key words for online indexing: Heart Aging Transcriptomics Proteomics eQTL pQTL Stoichiometry ABSTRACT Investigation of the molecular mechanisms of aging in the human heart is challenging due to confounding factors, such as diet and medications, as well limited access to tissues. The laboratory mouse provides an ideal model to study aging in healthy individuals in a controlled environment. However, previous mouse studies have examined only a narrow range of the genetic variation that shapes individual differences during aging. Here, we analyzed transcriptome and proteome data from hearts of genetically diverse mice at ages 6, 12 and 18 months to characterize molecular changes that occur in the aging heart. Transcripts and proteins reveal distinct biological processes that are altered through the course of natural aging. Transcriptome analysis reveals a scenario of cardiac hypertrophy, fibrosis, and reemergence of fetal gene expression patterns. -
Supplementary Figures and Tables
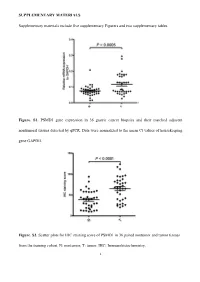
SUPPLEMENTARY MATERIALS Supplementary materials include five supplementary Figurers and two supplementary tables. Figure. S1. PSMD1 gene expression in 36 gastric cancer biopsies and their matched adjacent nontumoral tissues detected by qPCR. Data were normalized to the mean Ct values of housekeeping gene GAPDH. Figure. S2. Scatter plots for IHC staining score of PSMD1 in 36 paired nontumor and tumor tissues from the training cohort. N: nontumor; T: tumor. IHC: Immunohistochemistry. 1 Figure. S3. Kaplan-Meier survival curve analysis of DFS for all 411 patients with gastric cancer according to the PSMD1 expression stratified by clinicopathological risk factors. P-values were calculated by the log-rank test. 2 Figure. S4. Kaplan-Meier survival curve analysis of OS for all 411 patients with gastric cancer according to the PSMD1 expression stratified by clinicopathological risk factors. P-values were calculated by the log-rank test. 3 Figure. S5. Extended model for prediction of DFS and OS in patients with GC based on PSMD1 expression. ROC analyses of sensitivity and specificity for prediction of a, b DFS and c, d OS by the combined model (TNM stage and PSMD1 expression combination), TNM stage model, andPSMD1 expression model. 4 Figure. S6. X-tile plots of the DFS and OS nomograms. Color intensity of the plot represents low (dark, black) to high (bright, red, or green) strength of association at each division. Red represents the inverse association between the PSMD1 expression and survival (DFS or OS), whereas green represents direct association. a DFS nomogram, b OS nomogram. 5 Table S1. Clinical characteristics of the 36 patients with gastric cancer.