EZH1/2 Inhibitors Favor ILC3 Development from Human HSPC-CD34+ Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

HLA Mismatching Favoring Host-Versus-Graft NK Cell Activity Via KIR3DL1 Is Associated With
HLA mismatching favoring host-versus-graft NK cell activity via KIR3DL1 is associated with improved outcomes following lung transplantation John R. Greenland1,2,6, Haibo Sun3, Daniel Calabrese2, Tiffany Chong2, Jonathan P. Singer2, Jasleen Kukreja4, Steven R. Hays2, Jeffrey A. Golden2,4, George H. Caughey1,2,5, Jeffrey M. Venstrom2, Raja Rajalinginam3 1 Medical Service, Veterans Affairs Medical Center, San Francisco CA, 94121 2 Department of Medicine, University of California, San Francisco CA, 94143 3 Immunogenetics and Transplantation Laboratory, Department of Surgery, University of California, San Francisco CA, 94143 4 Department of Surgery, University of California, San Francisco CA, 94143 5 Cardiovascular Research Institute, University of California, San Francisco CA, 94143 6 Corresponding author: [email protected] Running Title: HVG NK cell activity in lung transplantation Abbreviations: antigen presenting cell (APC), bronchiolitis obliterans syndrome (BOS), bronchoalveolar lavage (BAL), chronic lung allograft dysfunction (CLAD), confidence interval (CI), forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), hazard ratio (HR), host-versus-graft (HVG), Killer cell immunoglobulin-like receptor (KIR), Major Histocompatibility Complex (MHC), Natural Killer (NK), restrictive allograft syndrome (RAS), United Network for Organ Sharing (UNOS), University of California, San Francisco (UCSF) Abstract Chronic lung allograft dysfunction (CLAD) is linked to rejection and limits survival following lung transplantation. HLA-Bw4 recipients of HLA-Bw6 grafts have enhanced host-versus-graft (HVG) NK cell activity mediated by KIR3DL1 ligand. Because natural killer (NK) cells may promote tolerance by depleting antigen-presenting cells, we hypothesized improved outcomes for HLA-Bw4 recipients of HLA- Bw6 grafts. We evaluated differences in acute cellular rejection (ACR) and CLAD-free survival across 252 KIR3DL1+ recipients from UCSF. -

Phyre 2 Results for A1YIY0
Email [email protected] Description A1YIY0 Tue Jul 30 13:19:15 BST Date 2013 Unique Job 1035bc4b501530df ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:cell adhesion Chain: A: PDB Molecule:down syndrome cell adhesion molecule 1 c3dmkA_ Alignment 100.0 14 (dscam) isoform PDBTitle: crystal structure of down syndrome cell adhesion molecule (dscam)2 isoform 1.30.30, n-terminal eight ig domains PDB header:cell adhesion 2 c2om5A_ Alignment 100.0 20 Chain: A: PDB Molecule:contactin 2; PDBTitle: n-terminal fragment of human tax1 PDB header:cell adhesion 3 c3jxaA_ Alignment 100.0 21 Chain: A: PDB Molecule:contactin 4; PDBTitle: immunoglobulin domains 1-4 of mouse cntn4 PDB header:signaling protein/transferase Chain: A: PDB Molecule:tek tyrosine kinase variant; 4 c4k0vA_ 100.0 12 Alignment PDBTitle: structural basis for angiopoietin-1 mediated signaling initiation PDB header:immune system Chain: A: PDB Molecule:natural cytotoxicity triggering receptor 1; 5 c1p6fA_ 100.0 9 Alignment PDBTitle: structure of the human natural cytotoxicity receptor nkp46 PDB header:immune system/receptor 6 c1ollA_ Alignment 99.9 9 Chain: A: PDB Molecule:nk receptor; PDBTitle: extracellular region of the human receptor nkp46 PDB header:viral protein Chain: A: PDB Molecule:hoc head outer capsid protein; 7 c3shsA_ 99.9 18 Alignment PDBTitle: three n-terminal domains of the bacteriophage rb49 highly immunogenic2 outer capsid protein (hoc) PDB header:immune system Chain: E: PDB Molecule:natural -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Snipa Snpcard
SNiPAcard Block annotations Block info genomic range chr19:55,117,749-55,168,602 block size 50,854 bp variant count 74 variants Basic features Conservation/deleteriousness Linked genes μ = -0.557 [-4.065 – AC009892.10 , AC009892.5 , AC009892.9 , AC245036.1 , AC245036.2 , phyloP 2.368] gene(s) hit or close-by AC245036.3 , AC245036.4 , AC245036.5 , AC245036.6 , LILRA1 , LILRB1 , LILRB4 , MIR8061 , VN1R105P phastCons μ = 0.059 [0 – 0.633] eQTL gene(s) CTB-83J4.2 , LILRA1 , LILRA2 , LILRB2 , LILRB5 , LILRP1 μ = -0.651 [-4.69 – 2.07] AC008984.5 , AC008984.5 , AC008984.6 , AC008984.6 , AC008984.7 , AC008984.7 , AC009892.10 , AC009892.10 , AC009892.2 , AC009892.2 , AC009892.5 , AC010518.3 , AC010518.3 , AC011515.2 , AC011515.2 , AC012314.19 , AC012314.19 , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , -
Type of the Paper (Article
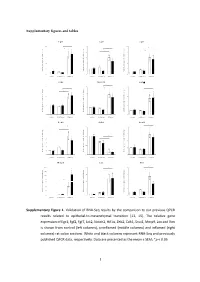
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

By Macaque NK Cells Killer Cell Ig-Like Receptor 3DL Expressed
Degenerate Recognition of MHC Class I Molecules with Bw4 and Bw6 Motifs by a Killer Cell Ig-like Receptor 3DL Expressed by Macaque NK Cells This information is current as of September 29, 2021. Sebastien M. Maloveste, Dan Chen, Emma Gostick, Julian P. Vivian, Ronald J. Plishka, Ranjini Iyengar, Robin L. Kruthers, Alicia Buckler-White, Andrew G. Brooks, Jamie Rossjohn, David A. Price and Bernard A. P. Lafont J Immunol 2012; 189:4338-4348; Prepublished online 5 Downloaded from October 2012; doi: 10.4049/jimmunol.1201360 http://www.jimmunol.org/content/189/9/4338 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2012/10/05/jimmunol.120136 Material 0.DC1 References This article cites 55 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/189/9/4338.full#ref-list-1 by guest on September 29, 2021 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -

KIR3DL1 and HLA-A and HLA-B Peptide-Dependent Interactions Between Cutting Edge: Allele-Specific
Cutting Edge: Allele-Specific and Peptide-Dependent Interactions between KIR3DL1 and HLA-A and HLA-B This information is current as Hathairat Thananchai, Geraldine Gillespie, Maureen P. of September 29, 2021. Martin, Arman Bashirova, Nobuyo Yawata, Makoto Yawata, Philippa Easterbrook, Daniel W. McVicar, Katsumi Maenaka, Peter Parham, Mary Carrington, Tao Dong and Sarah Rowland-Jones J Immunol 2007; 178:33-37; ; Downloaded from doi: 10.4049/jimmunol.178.1.33 http://www.jimmunol.org/content/178/1/33 References This article cites 28 articles, 12 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/178/1/33.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 29, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. THE JOURNAL OF IMMUNOLOGY CUTTING EDGE Cutting Edge: Allele-Specific and Peptide-Dependent Interactions between KIR3DL1 and HLA-A and HLA-B1,2 † ‡ Hathairat Thananchai,* Geraldine Gillespie,* Maureen P. -

KRAS Mutations Are Negatively Correlated with Immunity in Colon Cancer
www.aging-us.com AGING 2021, Vol. 13, No. 1 Research Paper KRAS mutations are negatively correlated with immunity in colon cancer Xiaorui Fu1,2,*, Xinyi Wang1,2,*, Jinzhong Duanmu1, Taiyuan Li1, Qunguang Jiang1 1Department of Gastrointestinal Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People's Republic of China 2Queen Mary College, Medical Department, Nanchang University, Nanchang, Jiangxi, People's Republic of China *Equal contribution Correspondence to: Qunguang Jiang; email: [email protected] Keywords: KRAS mutations, immunity, colon cancer, tumor-infiltrating immune cells, inflammation Received: March 27, 2020 Accepted: October 8, 2020 Published: November 26, 2020 Copyright: © 2020 Fu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The heterogeneity of colon cancer tumors suggests that therapeutics targeting specific molecules may be effective in only a few patients. It is therefore necessary to explore gene mutations in colon cancer. In this study, we obtained colon cancer samples from The Cancer Genome Atlas, and the International Cancer Genome Consortium. We evaluated the landscape of somatic mutations in colon cancer and found that KRAS mutations, particularly rs121913529, were frequent and had prognostic value. Using ESTIMATE analysis, we observed that the KRAS-mutated group had higher tumor purity, lower immune score, and lower stromal score than the wild- type group. Through single-sample Gene Set Enrichment Analysis and Gene Set Enrichment Analysis, we found that KRAS mutations negatively correlated with enrichment levels of tumor infiltrating lymphocytes, inflammation, and cytolytic activities. -

NK Cells: Receptors and Functions Eric Vivier and Sophie Ugolini
NK cells: receptors and functions Eric Vivier and Sophie Ugolini Natural killer (NK) cells were identified in 1975 as lymphocytes of the innate many processes that were originally restricted to adaptive immunity, such immune system that can kill tumour cells. Since then, NK cells have been as priming, education and memory, are now known to occur in NK cells. shown to kill an array of ‘stressed’ cells and secrete cytokines that Indeed, NK cells undergo sophisticated processes of adaptation that allow participate in shaping adaptive immune responses. A key feature of NK cells them to be tuned to their environment. There is also a growing interest in resides in their capacity to distinguish stressed cells (such as tumour cells, manipulating NK cells in innovative therapeutic settings. For example, the infected cells and damaged cells) from normal cells. Although NK cells are understanding of NK cell inhibition by MHC class I-specific receptors has generally considered to be components of early innate immune defence, prompted the design of innovative anticancer therapies. The NK cell detection system includes numerous receptors, Key receptors on human NK cells the engagement of which dictates the quality and intensity Inhibitory receptors Activating receptors, adhesion molecules or co-stimulatory molecules of the NK cell response. NK cells use inhibitory receptors to gauge the absence of constitutively expressed self molecules Target cell Nectin 2 SLAMF7 Sialic CLEC2D (CD112, PVRL2) NECL2 L-selectin NECL5 AICL NTB-A (CRACC, on susceptible target cells. As a consequence, NK cells can Receptors for MHC class I and class I-Iike molecules B7-H6 acid Cadherins (LLT1) CMV pp65 NECL5 (CADM1) (CD62L) (CD155, PVR) (CLEC2B) CEACAM1 (SLAMF6) CS1, CD319) recognize ‘missing self’ on haematopoietic cells. -

KIR3DL1/S1 Receptor on Human NK Cells Functional Polymorphism Of
Functional Polymorphism of the KIR3DL1/S1 Receptor on Human NK Cells Geraldine M. O'Connor, Kieran J. Guinan, Rodat T. Cunningham, Derek Middleton, Peter Parham and Clair M. This information is current as Gardiner of October 2, 2021. J Immunol 2007; 178:235-241; ; doi: 10.4049/jimmunol.178.1.235 http://www.jimmunol.org/content/178/1/235 Downloaded from References This article cites 28 articles, 10 of which you can access for free at: http://www.jimmunol.org/content/178/1/235.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Functional Polymorphism of the KIR3DL1/S1 Receptor on Human NK Cells1 Geraldine M. O’Connor,* Kieran J. Guinan,* Rodat T. Cunningham,† Derek Middleton,† Peter Parham,‡ and Clair M. Gardiner2* NK cells express both inhibitory and activatory receptors that allow them to recognize target cells through HLA class I Ag expression. -

Genetics of Microscopic Polyangiitis in the Japanese Population
Ann Vasc Dis Vol.5, No.3; 2012; pp 289–295 ©2012 Annals of Vascular Diseases doi: 10.3400/avd.ra.12.00062 Review Article Genetics of Microscopic Polyangiitis in the Japanese Population Naoyuki Tsuchiya, MD, PhD The epidemiology of ANCA-associated vasculitis is substantially different between Caucasians and Japanese, which may be related to differences in genetic backgrounds. In this review, I discussed our findings on the genetics of microscopic polyangiitis (MPA) in Japanese. Analysis of HLA genes revealed a significant increase in the HLA-DRB1*09:01-DQB1*03:03 haplotype MPA. This is one of the most frequent haplotypes in Japanese, but is nearly absent in Caucasians, and has been shown to be associated with multiple autoimmune diseases. Analysis of KIR genes revealed significant decreases in the carrier frequency of an activating receptor KIR2DS3 in MPA. When KIRs were analyzed in combination with HLA ligands, the proportion of individuals carrying KIR3DL1 and HLA-Bw4 but not KIR3DS1, the most inhibitory of all KIR3DS1/3DL1/ HLA-B combinations, was significantly increased in MPA. These results suggested that decreased activation of NK and/or T cells may cause a predisposition to MPA. LILRA2 is an activating receptor involved in granulocyte and macrophage activation. LILRA2 SNP rs2241524 G >A, which disrupts the intron 6 splice acceptor site, was significantly associated with MPA. The risk allele produces an LILRA2 isoform lacking three amino acids in the linker region. These findings, when confirmed by larger-scale studies, will shed light on the molecular mechanisms of MPA. (*English Translation of J Jpn Coll Angiol 2009; 49: 31-37.) Keywords: microscopic polyangiitis, genetics, HLA, KIR, LILRA2 INTRODUCTION populations are associated with certain genetic and en- vironmental factors. -

KIR) 3DL1 Down-Regulation Enhances Inhibition of Type 1 Diabetes by Autoantigen-Specific Regulatory T Cells
Killer cell Ig-like receptor (KIR) 3DL1 down-regulation enhances inhibition of type 1 diabetes by autoantigen-specific regulatory T cells Hanjun Qina,b, Zunde Wangb,1, Weiting Dua,b, Wen-Hui Leea,b, Xiwei Wuc, Arthur D. Riggsb,2, and Chih-Pin Liua,b,2 Departments of aImmunology, bDiabetes and Metabolic Disease Research, and cMolecular Medicine, The Beckman Research Institute of the City of Hope, Duarte, CA 91010-3000 Contributed by Arthur D. Riggs, December 21, 2010 (sent for review November 5, 2010) Both Foxp3+ regulatory T cells (Tregs) and antigen-expanded To identify factors that may be involved in modulating Treg − Foxp3 Tregs play an important role in regulating immune function, we performed genome-wide comparative expression responses as well as in preventing autoimmune diseases and graft analyses of CD4+ Tregs, Tpaths, and non-Tpaths and noted rejection. Molecular mechanisms modulating Treg function remain unique expression of killer cell Ig-like receptor (KIR) 3DL1 in largely unclear, however. We report here on the expression and Tregs. KIRs are transmembrane glycoproteins expressed by nat- function of an inhibitory killer cell Ig-like receptor, KIR3DL1, in ural killer (NK) cells. KIR genes have been studied mainly in a nonobese diabetic (NOD) mouse-derived autoantigen-specific humans, where they are known to be involved in self-nonself Treg (2D2), which protects from type 1 diabetes (T1D) in adoptive recognition by binding to ligands on target cells (13–19). For some transfer experiments. This gene is not expressed in T1D patho- KIRs, the ligand is an HLA class I membrane protein. By rec- genic T cells (Tpaths) or non-Tpath T cells.