KIR) 3DL1 Down-Regulation Enhances Inhibition of Type 1 Diabetes by Autoantigen-Specific Regulatory T Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

HLA Mismatching Favoring Host-Versus-Graft NK Cell Activity Via KIR3DL1 Is Associated With
HLA mismatching favoring host-versus-graft NK cell activity via KIR3DL1 is associated with improved outcomes following lung transplantation John R. Greenland1,2,6, Haibo Sun3, Daniel Calabrese2, Tiffany Chong2, Jonathan P. Singer2, Jasleen Kukreja4, Steven R. Hays2, Jeffrey A. Golden2,4, George H. Caughey1,2,5, Jeffrey M. Venstrom2, Raja Rajalinginam3 1 Medical Service, Veterans Affairs Medical Center, San Francisco CA, 94121 2 Department of Medicine, University of California, San Francisco CA, 94143 3 Immunogenetics and Transplantation Laboratory, Department of Surgery, University of California, San Francisco CA, 94143 4 Department of Surgery, University of California, San Francisco CA, 94143 5 Cardiovascular Research Institute, University of California, San Francisco CA, 94143 6 Corresponding author: [email protected] Running Title: HVG NK cell activity in lung transplantation Abbreviations: antigen presenting cell (APC), bronchiolitis obliterans syndrome (BOS), bronchoalveolar lavage (BAL), chronic lung allograft dysfunction (CLAD), confidence interval (CI), forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), hazard ratio (HR), host-versus-graft (HVG), Killer cell immunoglobulin-like receptor (KIR), Major Histocompatibility Complex (MHC), Natural Killer (NK), restrictive allograft syndrome (RAS), United Network for Organ Sharing (UNOS), University of California, San Francisco (UCSF) Abstract Chronic lung allograft dysfunction (CLAD) is linked to rejection and limits survival following lung transplantation. HLA-Bw4 recipients of HLA-Bw6 grafts have enhanced host-versus-graft (HVG) NK cell activity mediated by KIR3DL1 ligand. Because natural killer (NK) cells may promote tolerance by depleting antigen-presenting cells, we hypothesized improved outcomes for HLA-Bw4 recipients of HLA- Bw6 grafts. We evaluated differences in acute cellular rejection (ACR) and CLAD-free survival across 252 KIR3DL1+ recipients from UCSF. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Expression Tissue
The Journal of Immunology Killer Cell Ig-Like Receptor and Leukocyte Ig-Like Receptor Transgenic Mice Exhibit Tissue- and Cell-Specific Transgene Expression1 Danny Belkin,* Michaela Torkar,* Chiwen Chang,* Roland Barten,* Mauro Tolaini,† Anja Haude,* Rachel Allen,* Michael J. Wilson,* Dimitris Kioussis,† and John Trowsdale2* To generate an experimental model for exploring the function, expression pattern, and developmental regulation of human Ig-like activating and inhibitory receptors, we have generated transgenic mice using two human genomic clones: 52N12 (a 150-Kb clone encompassing the leukocyte Ig-like receptor (LILR)B1 (ILT2), LILRB4 (ILT3), and LILRA1 (LIR6) genes) and 1060P11 (a 160-Kb clone that contains ten killer cell Ig-like receptor (KIR) genes). Both the KIR and LILR families are encoded within the leukocyte receptor complex, and are involved in immune modulation. We have also produced a novel mAb to LILRA1 to facilitate expression studies. The LILR transgenes were expressed in a similar, but not identical, pattern to that observed in humans: LILRB1 was expressed in B cells, most NK cells, and a small number of T cells; LILRB4 was expressed in a B cell subset; and LILRA1 was found on a ring of cells surrounding B cell areas on spleen sections, consistent with other data showing monocyte/macrophage expression. KIR transgenic mice showed KIR2DL2 expression on a subset of NK cells and T cells, similar to the pattern seen in humans, and expression of KIR2DL4, KIR3DS1, and KIR2DL5 by splenic NK cells. These observations indicate that linked regulatory elements within the genomic clones are sufficient to allow appropriate expression of KIRs in mice, and illustrate that the presence of the natural ligands for these receptors, in the form of human MHC class I proteins, is not necessary for the expression of the KIRs observed in these mice. -

Phyre 2 Results for A1YIY0
Email [email protected] Description A1YIY0 Tue Jul 30 13:19:15 BST Date 2013 Unique Job 1035bc4b501530df ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:cell adhesion Chain: A: PDB Molecule:down syndrome cell adhesion molecule 1 c3dmkA_ Alignment 100.0 14 (dscam) isoform PDBTitle: crystal structure of down syndrome cell adhesion molecule (dscam)2 isoform 1.30.30, n-terminal eight ig domains PDB header:cell adhesion 2 c2om5A_ Alignment 100.0 20 Chain: A: PDB Molecule:contactin 2; PDBTitle: n-terminal fragment of human tax1 PDB header:cell adhesion 3 c3jxaA_ Alignment 100.0 21 Chain: A: PDB Molecule:contactin 4; PDBTitle: immunoglobulin domains 1-4 of mouse cntn4 PDB header:signaling protein/transferase Chain: A: PDB Molecule:tek tyrosine kinase variant; 4 c4k0vA_ 100.0 12 Alignment PDBTitle: structural basis for angiopoietin-1 mediated signaling initiation PDB header:immune system Chain: A: PDB Molecule:natural cytotoxicity triggering receptor 1; 5 c1p6fA_ 100.0 9 Alignment PDBTitle: structure of the human natural cytotoxicity receptor nkp46 PDB header:immune system/receptor 6 c1ollA_ Alignment 99.9 9 Chain: A: PDB Molecule:nk receptor; PDBTitle: extracellular region of the human receptor nkp46 PDB header:viral protein Chain: A: PDB Molecule:hoc head outer capsid protein; 7 c3shsA_ 99.9 18 Alignment PDBTitle: three n-terminal domains of the bacteriophage rb49 highly immunogenic2 outer capsid protein (hoc) PDB header:immune system Chain: E: PDB Molecule:natural -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Flow Reagents Single Color Antibodies CD Chart
CD CHART CD N° Alternative Name CD N° Alternative Name CD N° Alternative Name Beckman Coulter Clone Beckman Coulter Clone Beckman Coulter Clone T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells CD1a T6, R4, HTA1 Act p n n p n n S l CD99 MIC2 gene product, E2 p p p CD223 LAG-3 (Lymphocyte activation gene 3) Act n Act p n CD1b R1 Act p n n p n n S CD99R restricted CD99 p p CD224 GGT (γ-glutamyl transferase) p p p p p p CD1c R7, M241 Act S n n p n n S l CD100 SEMA4D (semaphorin 4D) p Low p p p n n CD225 Leu13, interferon induced transmembrane protein 1 (IFITM1). p p p p p CD1d R3 Act S n n Low n n S Intest CD101 V7, P126 Act n p n p n n p CD226 DNAM-1, PTA-1 Act n Act Act Act n p n CD1e R2 n n n n S CD102 ICAM-2 (intercellular adhesion molecule-2) p p n p Folli p CD227 MUC1, mucin 1, episialin, PUM, PEM, EMA, DF3, H23 Act p CD2 T11; Tp50; sheep red blood cell (SRBC) receptor; LFA-2 p S n p n n l CD103 HML-1 (human mucosal lymphocytes antigen 1), integrin aE chain S n n n n n n n l CD228 Melanotransferrin (MT), p97 p p CD3 T3, CD3 complex p n n n n n n n n n l CD104 integrin b4 chain; TSP-1180 n n n n n n n p p CD229 Ly9, T-lymphocyte surface antigen p p n p n -

Detection of Pro Angiogenic and Inflammatory Biomarkers in Patients With
www.nature.com/scientificreports OPEN Detection of pro angiogenic and infammatory biomarkers in patients with CKD Diana Jalal1,2,3*, Bridget Sanford4, Brandon Renner5, Patrick Ten Eyck6, Jennifer Laskowski5, James Cooper5, Mingyao Sun1, Yousef Zakharia7, Douglas Spitz7,9, Ayotunde Dokun8, Massimo Attanasio1, Kenneth Jones10 & Joshua M. Thurman5 Cardiovascular disease (CVD) is the most common cause of death in patients with native and post-transplant chronic kidney disease (CKD). To identify new biomarkers of vascular injury and infammation, we analyzed the proteome of plasma and circulating extracellular vesicles (EVs) in native and post-transplant CKD patients utilizing an aptamer-based assay. Proteins of angiogenesis were signifcantly higher in native and post-transplant CKD patients versus healthy controls. Ingenuity pathway analysis (IPA) indicated Ephrin receptor signaling, serine biosynthesis, and transforming growth factor-β as the top pathways activated in both CKD groups. Pro-infammatory proteins were signifcantly higher only in the EVs of native CKD patients. IPA indicated acute phase response signaling, insulin-like growth factor-1, tumor necrosis factor-α, and interleukin-6 pathway activation. These data indicate that pathways of angiogenesis and infammation are activated in CKD patients’ plasma and EVs, respectively. The pathways common in both native and post-transplant CKD may signal similar mechanisms of CVD. Approximately one in 10 individuals has chronic kidney disease (CKD) rendering CKD one of the most common diseases worldwide1. CKD is associated with a high burden of morbidity in the form of end stage kidney disease (ESKD) requiring dialysis or transplantation 2. Furthermore, patients with CKD are at signifcantly increased risk of death from cardiovascular disease (CVD)3,4. -

Differential Integrin Adhesome Expression Defines Human Natural
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.01.404806; this version posted December 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Differential integrin adhesome expression defines human natural killer cell residency and developmental stage Everardo Hegewisch Solloa1, Seungmae Seo1, Bethany L. Mundy-Bosse2, Anjali Mishra3,a, Erik Waldman4,b, Sarah Maurrasse4,b, Eli Grunstein4, Thomas J. Connors5, Aharon G. Freud2,6, and Emily M. Mace1 1Department of Pediatrics, Columbia University College of Physicians and Surgeons, New York NY 10032 2Division of Hematology, Department of Internal Medicine, The Ohio State University, Columbus, OH 43210, USA; Comprehensive Cancer Center and The James Cancer Hospital and Solove Research Institute, The Ohio State University, Columbus, OH 43210 33Division of Dermatology, Department of Internal Medicine, The Ohio State University, Columbus, OH 43210, USA; Comprehensive Cancer Center and The James Cancer Hospital and Solove Research Institute, The Ohio State University, Columbus, OH 43210 4Department of Otolaryngology - Head and Neck Surgery, Columbia University Medical Center, New York, New York 10032 5Department of Pediatrics, Division of Pediatric Critical Care and Hospital Medicine, Columbia University Irving Medical Center, New York, NY 10024 6Department of Pathology, The Ohio State University, Columbus, -

Snipa Snpcard
SNiPAcard Block annotations Block info genomic range chr19:55,117,749-55,168,602 block size 50,854 bp variant count 74 variants Basic features Conservation/deleteriousness Linked genes μ = -0.557 [-4.065 – AC009892.10 , AC009892.5 , AC009892.9 , AC245036.1 , AC245036.2 , phyloP 2.368] gene(s) hit or close-by AC245036.3 , AC245036.4 , AC245036.5 , AC245036.6 , LILRA1 , LILRB1 , LILRB4 , MIR8061 , VN1R105P phastCons μ = 0.059 [0 – 0.633] eQTL gene(s) CTB-83J4.2 , LILRA1 , LILRA2 , LILRB2 , LILRB5 , LILRP1 μ = -0.651 [-4.69 – 2.07] AC008984.5 , AC008984.5 , AC008984.6 , AC008984.6 , AC008984.7 , AC008984.7 , AC009892.10 , AC009892.10 , AC009892.2 , AC009892.2 , AC009892.5 , AC010518.3 , AC010518.3 , AC011515.2 , AC011515.2 , AC012314.19 , AC012314.19 , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , -
Type of the Paper (Article
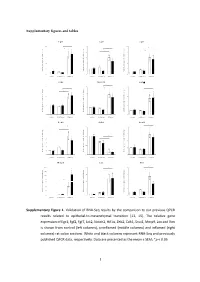
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients with Stable Coronary Heart Disease
Supplementary Online Content Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. doi: 10.1001/jama.2016.5951 eTable 1. List of 1130 Proteins Measured by Somalogic’s Modified Aptamer-Based Proteomic Assay eTable 2. Coefficients for Weibull Recalibration Model Applied to 9-Protein Model eFigure 1. Median Protein Levels in Derivation and Validation Cohort eTable 3. Coefficients for the Recalibration Model Applied to Refit Framingham eFigure 2. Calibration Plots for the Refit Framingham Model eTable 4. List of 200 Proteins Associated With the Risk of MI, Stroke, Heart Failure, and Death eFigure 3. Hazard Ratios of Lasso Selected Proteins for Primary End Point of MI, Stroke, Heart Failure, and Death eFigure 4. 9-Protein Prognostic Model Hazard Ratios Adjusted for Framingham Variables eFigure 5. 9-Protein Risk Scores by Event Type This supplementary material has been provided by the authors to give readers additional information about their work. Downloaded From: https://jamanetwork.com/ on 10/02/2021 Supplemental Material Table of Contents 1 Study Design and Data Processing ......................................................................................................... 3 2 Table of 1130 Proteins Measured .......................................................................................................... 4 3 Variable Selection and Statistical Modeling ........................................................................................ -

Natural Killer Cell Lymphoma Shares Strikingly Similar Molecular Features
Leukemia (2011) 25, 348–358 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu ORIGINAL ARTICLE Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic cd T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro J Iqbal1, DD Weisenburger1, A Chowdhury2, MY Tsai2, G Srivastava3, TC Greiner1, C Kucuk1, K Deffenbacher1, J Vose4, L Smith5, WY Au3, S Nakamura6, M Seto6, J Delabie7, F Berger8, F Loong3, Y-H Ko9, I Sng10, X Liu11, TP Loughran11, J Armitage4 and WC Chan1, for the International Peripheral T-cell Lymphoma Project 1Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE, USA; 2Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA; 3Departments of Pathology and Medicine, University of Hong Kong, Queen Mary Hospital, Hong Kong, China; 4Division of Hematology and Oncology, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA; 5College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA; 6Departments of Pathology and Cancer Genetics, Aichi Cancer Center Research Institute, Nagoya University, Nagoya, Japan; 7Department of Pathology, University of Oslo, Norwegian Radium Hospital, Oslo, Norway; 8Department of Pathology, Centre Hospitalier Lyon-Sud, Lyon, France; 9Department of Pathology, Samsung Medical Center, Sungkyunkwan University, Seoul, Korea; 10Department of Pathology, Singapore General Hospital, Singapore and 11Penn State Hershey Cancer Institute, Pennsylvania State University College of Medicine, Hershey, PA, USA Natural killer (NK) cell lymphomas/leukemias are rare neo- Introduction plasms with an aggressive clinical behavior.