Anti-Vascular Endothelial Growth Factor Receptor-1Antagonist
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1: Clinical Pharmacokinetics 1
1: CLINICAL PHARMACOKINETICS 1 General overview: clinical pharmacokinetics, 2 Pharmacokinetics, 4 Drug clearance (CL), 6 Volume of distribution (Vd), 8 The half-life (t½), 10 Oral availability (F), 12 Protein binding (PB), 14 pH and pharmacokinetics, 16 1 Clinical pharmacokinetics General overview General overview: clinical pharmacokinetics 1 The ultimate aim of drug therapy is to achieve effi cacy without toxicity. This involves achieving a plasma concentration (Cp) within the ‘therapeutic window’, i.e. above the min- imal effective concentration (MEC), but below the minimal toxic concentration (MTC). Clinical pharmacokinetics is about all the factors that determine variability in the Cp and its time-course. The various factors are dealt with in subsequent chapters. Ideal therapeutics: effi cacy without toxicity Minimum Toxic Concentration (MTC) Ideal dosing Minimum Effective Concentration (MEC) Drug concentration Time The graph shows a continuous IV infusion at steady state, where the dose-rate is exactly appropriate for the patient’s clearance (CL). Inappropriate dosing Dosing too high in relation to the patient’s CL – toxicity likely Minimum Toxic Concentration (MTC) Minimum Effective Concentration (MEC) Dosing too low in relation to the Drug concentration patient’s CL – drug may be ineffective Time Some reasons for variation in CL Low CL High CL Normal variation Normal variation Renal impairment Increased renal blood fl ow Genetic poor metabolism Genetic hypermetabolism Liver impairment Enzyme induction Enzyme inhibition Old age/neonate 2 General overview Clinical Pharmacokinetics Pharmacokinetic factors determining ideal therapeutics If immediate effect is needed, a loading dose (LD) must be given to achieve a desired 1 concentration. The LD is determined by the volume of distribution (Vd). -

CPIC Guideline for Pharmacogenetics-Guided Warfarin Dosing – Supplement V2.0 1 Table of Contents Guideline Updates
Supplement to: Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for Pharmacogenetics-guided Warfarin Dosing: 2016 Update Julie A. Johnson1, Kelly Caudle2, Li Gong3, Michelle Whirl-Carrillo3, C. Michael Stein4, Stuart A. Scott5, Ming Ta Michael Lee6 , Brian F. Gage7, Stephen E. Kimmel8,9, Minoli A. Perera10, Jeffrey L. Anderson11, Munir Pirmohamed12, Teri E. Klein3, Nita A. Limdi13, Larisa H. Cavallari1, Mia Wadelius14 1Department of Pharmacotherapy and Translational Research, College of Pharmacy, and Center for Pharmacogenomics, University of Florida, Gainesville, Florida, USA 2Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN 3Department of Genetics, Stanford University, Stanford, California, USA 4Division of Clinical Pharmacology Vanderbilt Medical School, Nashville, TN, USA 5Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA 6Laboratory for International Alliance on Genomic Research, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan; National Center for Genome Medicine; Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan; Genomic Medicine Institute Geisinger Health system, Danville, PA 7Department of Internal Medicine, Washington University in St. Louis, St. Louis, Missouri 8Center for Clinical Epidemiology and Biostatistics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA 9Department of Medicine and Department of Biostatistics and Epidemiology, University of Pennsylvania -

Efavirenz) Capsules and Tablets 3 Rx Only
1 SUSTIVA® 2 (efavirenz) capsules and tablets 3 Rx only 4 DESCRIPTION 5 SUSTIVA® (efavirenz) is a human immunodeficiency virus type 1 (HIV-1) specific, non- 6 nucleoside, reverse transcriptase inhibitor (NNRTI). 7 Capsules: SUSTIVA is available as capsules for oral administration containing either 8 50 mg, 100 mg, or 200 mg of efavirenz and the following inactive ingredients: lactose 9 monohydrate, magnesium stearate, sodium lauryl sulfate, and sodium starch glycolate. 10 The capsule shell contains the following inactive ingredients and dyes: gelatin, sodium 11 lauryl sulfate, titanium dioxide, and/or yellow iron oxide. The capsule shells may also 12 contain silicon dioxide. The capsules are printed with ink containing carmine 40 blue, 13 FD&C Blue No. 2, and titanium dioxide. 14 Tablets: SUSTIVA is available as film-coated tablets for oral administration containing 15 600 mg of efavirenz and the following inactive ingredients: croscarmellose sodium, 16 hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, microcrystalline 17 cellulose, and sodium lauryl sulfate. The film coating contains Opadry® Yellow and 18 Opadry® Clear. The tablets are polished with carnauba wax and printed with purple ink, 19 Opacode® WB. 20 Efavirenz is chemically described as (S)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4- 21 (trifluoromethyl)-2H-3,1-benzoxazin-2-one. 22 Its empirical formula is C14H9ClF3NO2 and its structural formula is: 1 of 45 Approved v2.0 F C 3 Cl O NO 23 H 24 Efavirenz is a white to slightly pink crystalline powder with a molecular mass of 315.68. 25 It is practically insoluble in water (<10 µg/mL). -
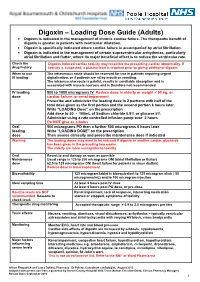
Digoxin – Loading Dose Guide (Adults) Digoxin Is Indicated in the Management of Chronic Cardiac Failure
Digoxin – Loading Dose Guide (Adults) Digoxin is indicated in the management of chronic cardiac failure. The therapeutic benefit of digoxin is greater in patients with ventricular dilatation. Digoxin is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Digoxin is indicated in the management of certain supraventricular arrhythmias, particularly atrial fibrillation and flutter, where its major beneficial effect is to reduce the ventricular rate. Check the Digoxin-induced cardiac toxicity may resemble the presenting cardiac abnormality. If drug history toxicity is suspected, a plasma level is required prior to giving additional digoxin. When to use The intravenous route should be reserved for use in patients requiring urgent IV loading digitalisation, or if patients are nil by mouth or vomiting. The intramuscular route is painful, results in unreliable absorption and is associated with muscle necrosis and is therefore not recommended. IV loading 500 to 1000 micrograms IV Reduce dose in elderly or weight < 50 kg, or dose cardiac failure, or renal impairment Prescribe and administer the loading dose in 2 portions with half of the total dose given as the first portion and the second portion 6 hours later. Write “LOADING Dose” on the prescription Add dose to 50 - 100mL of Sodium chloride 0.9% or glucose 5% Administer using a rate controlled infusion pump over 2 hours Do NOT give as a bolus Oral 500 micrograms PO then a further 500 micrograms 6 hours later loading Write “LOADING DOSE” on the prescription dose Then assess clinically and prescribe maintenance dose if indicated Warning The loading doses may need to be reduced if digoxin or another cardiac glycoside has been given in the preceding two weeks. -

Guidelines for Use of Digoxin (Lanoxin )
Guidelines for use of Digoxin (Lanoxin) Recommended Neonatal Dose, Route, and Interval Loading or digitalizing dose: Total Loading Dose PMA (Weeks) IV (mcg/kg) PO (mcg/kg) 29 15 20 30 to 36 20 25 37 to 48 30 40 49 40 50 Divide into 3 doses over 24 hours Generally given in the following manner: 1. One-half the loading dose given immediately IV or PO 2. One-fourth the loading dose given 8 to 12 hours later IV or PO 3. The remaining one-fourth loading dose given after an additional 8 to 12 hours IV or PO 4. Administer IV slow push over 5 to 10 minutes 5. Obtain ECG 6 hours after digitalizing dose to assess for toxicity Maintenance dose: should be started 12 hours after the loading dose is completed Maintenance Dose PMA (Weeks) IV (mcg/kg) PO (mcg/kg) Interval (hours) 29 4 5 24 30 to 36 5 6 24 37 to 48 4 5 12 49 5 6 12 Titrate based on clinical response Chief Indications 1. Heart failure caused by diminished myocardial contractility 2. Supraventricular tachycardia, atrial flutter, atrial fibrillation Possible Adverse Reactions 1. Atrial or ventricular arrhythmias are common and may be an early indication of overdosage 2. Feeding intolerance, vomiting, diarrhea 3. Hypokalemia is associated with chronic Digoxin toxicity 4. Bradycardia due to depression of A-V conduction 5. Diuresis from improved cardiac output 6. Lethargy or seizures with toxicity Contraindications & Precautions 1. CAUTION use with pre-existing hypokalemia may lead to adverse reactions 2. CAUTION use with Indomethacin - may inhibit excretion of Digoxin 3. -

Perjeta Full Prescribing Information
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use PERJETA safely and effectively. See full prescribing information for --------------------- DOSAGE FORMS AND STRENGTHS---------------------- PERJETA. Injection: 420 mg/14 mL single-dose vial. (3) PERJETA® (pertuzumab) injection, for intravenous use ------------------------------ CONTRAINDICATIONS ------------------------------ Initial U.S. Approval: 2012 PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients. (4) WARNING: LEFT VENTRICULAR DYSFUNCTION and EMBRYO-FETAL TOXICITY ----------------------- WARNINGS AND PRECAUTIONS ----------------------- See full prescribing information for complete boxed warning. Infusion-Related Reactions: Monitor for signs and symptoms. If a Left Ventricular Dysfunction: PERJETA can result in significant infusion-associated reaction occurs, slow or interrupt the subclinical and clinical cardiac failure manifesting as decreased infusion and administer appropriate medical therapies. (5.3) LVEF and CHF. Evaluate cardiac function prior to and during Hypersensitivity Reactions/Anaphylaxis: Monitor for signs and treatment. Discontinue PERJETA treatment for a confirmed symptoms, including angioedema. If a severe hypersensitivity clinically significant decrease in left ventricular function. (2.3, reaction/anaphylaxis occurs, discontinue the infusion immediately and 5.1, 6.1) administer appropriate medical therapies. (5.4) Embryo-fetal Toxicity: Exposure to -

VANCOMYCIN DOSING and MONITORING GUIDELINES (NB Provincial Health Authorities Anti-Infective Stewardship Committee)
Amended: October 2020 VANCOMYCIN DOSING AND MONITORING GUIDELINES (NB Provincial Health Authorities Anti-Infective Stewardship Committee) GENERAL COMMENTS • Vancomycin is a glycopeptide antibiotic with bactericidal activity • It is active against gram-positive bacteria, including methicillin-resistant staphylococcus (MRSA) • Vancomycin is less effective than beta-lactams against Staphylococcus aureus that is susceptible to cloxacillin/methicillin • Vancomycin exhibits time-dependent killing: its effect depends primarily upon the time the concentration exceeds the organism’s Minimum Inhibitory Concentration (MIC) • These guidelines pertain to IV vancomycin only; they do not apply to PO vancomycin, which is not absorbed • Ensure that an adequate mg/kg dose and appropriate interval are ordered initially. Adjust the dose if necessary immediately; do not wait for a confirmatory trough level. • When managing a severe Staphylococcus aureus infection (e.g., bacteremia), an Infectious Diseases consultation is strongly encouraged. VANCOMYCIN IN ADULT PATIENTS ADULT INITIAL DOSE Loading dose: • Consider using a loading dose in patients with: o severe infections where rapid attainment of target level of 10-15 mg/mL is desired o significant renal dysfunction in order to decrease the time required to attain steady state • Recommended dose: 25-30 mg/kg IV o based on actual body weight, for 1 dose, followed by maintenance dose separated by recommended dosing interval o consider capping the loading dose at a maximum of 3g o loading doses DO NOT need to -

Loading Doses in Primary Care
Hull and East Riding Prescribing Committee Preventing fatalities from medication loading doses: Guidance for Primary Care Background A loading dose is an initial large dose of a medicine used to ensure a quick therapeutic response. It is usually given for a short period before therapy continues with a lower maintenance dose. The use of loading doses of medicines can be complex and error prone. Incorrect use of loading doses or subsequent maintenance regimens may lead to severe harm or death. The National Patient Safety Agency (NPSA) issued a Rapid Response Report (NPSA/2010/RRR018) in November 2010, with recommended actions aimed to reduce the number and severity of medication incidents involving incorrect prescribing or administration of loading doses and subsequent maintenance doses. These actions included production of a critical list of medicines, where incorrect loading doses or subsequent maintenance doses are likely to cause harm and ensuring healthcare professionals in primary care are aware of when to challenge abnormal doses of medicines on the agreed critical list. This guidance has been developed as a response to these recommended actions. Critical List Agreed critical list of drugs most likely to cause harm as a result of incorrect prescribing or administration of loading dose or subsequent maintenance dose are: Amiodarone Digoxin Phenytoin Warfarin Best practice in the prescribing, supply and administration of drugs requiring loading dose 1. Where a loading dose is prescribed or recommended, ensure that details of on- going treatment and titration to maintenance dose are clear, and in line with national or local guidelines. 2. Challenge any abnormal prescribing or treatment recommendations (see page 2). -

Premarket Evaluation in Early-Phase Clinical Studies and Recommendations for Labeling
Guidance for Industry Clinical Pharmacogenomics: Premarket Evaluation in Early-Phase Clinical Studies and Recommendations for Labeling U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Center for Devices and Radiological Health (CDRH) January 2013 Clinical Pharmacology Clinical/Medical 10300.fnl.doc Guidance for Industry Clinical Pharmacogenomics: Premarket Evaluation in Early-Phase Clinical Studies and Recommendations for Labeling Additional copies are available from: Office of Communications Division of Drug Information, WO51, Room 2201 10903 New Hampshire Ave. Silver Spring, MD 20993-0002 Phone: 301-796-3400; Fax 301-847-8714 http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm or Office of Communication, Outreach, and Development (HFM-40) Center for Biologics Evaluation and Research Food and Drug Administration 1401 Rockville Pike, Rockville, MD 20852-1448 http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (Tel) 800-835-4709 or 301-827-1800 or Office of Communication, Education, and Radiation Programs Division of Small Manufacturers, International and Consumer Assistance Center for Devices and Radiological Health Food and Drug Administration 10903 New Hampshire Ave. WO66, Room 4613 Silver Spring, MD 20993-0002 http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/default.htm Email: [email protected] Fax: 301-827-8149 (Tel) Manufacturers Assistance: 800-638-2041 or 301-796-7100 (Tel) International Staff: 301-796-5708 U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Center for Devices and Radiological Health (CDRH) January 2013 Clinical Pharmacology Clinical/Medical TABLE OF CONTENTS I. -

761065Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 761065Orig1s000 CLINICAL MICROBIOLOGY/VIROLOGY REVIEW(S) DIVISION OF ANTIVIRAL PRODUCTS CLINICAL VIROLOGY REVIEW BLA: 761065 SDN: 000 Addendum (Original BLA, SDN 012 in DARRTS) REVIEW COMPLETED: 01/19/2018 Clinical Virology Reviewer: Eric F. Donaldson, Ph.D. BLA#: 761065 SDN 000 (Original BLA) Reviewer Name(s): Eric F. Donaldson, Ph.D. Sponsor: TaiMed Biologics, Inc. 5251 California Ave., Suite 230 Irvine, CA 92617 Helen P. Shu, Ph.D. Vice President of Regulatory Affairs and Quality 858-481-6863 858-724-1844 (FAX) [email protected] Initial Complete Submission Dates: Correspondence Date: 5/3/2017 CDER Receipt Date: 5/3/2017 Assigned Date: 5/3/2017 Review Complete Date: 10/03/2017 PDUFA Date: 1/3/2018 extended to: 4/3/2018 Proprietary Name (Proposed): TROGARZO Drug Names: ibalizumab, TNX-355, Hu5A8, Mu5A8, TMB-355 Drug Class: CD4 post-attachment HIV-1 inhibitor Parent IND #: IND009776 Ibalizumab heavy chain primary structure (IgG4) QVQLQQSGPEVVKPGASVKMSCKASGYTFTSYVIHWVRQKPGQGLDWIGYINPYNDGTDYDEKFKGKATLTSDTSTSTAYM ELSSLRSEDTAVYYCAREKDNYATGAWFAYWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVS WNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPSCPAPEFLGGPSVF LFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCK VSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGS FFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK Ibalizumab light chain primary structure (κ) DIVMTQSPDSLAVSLGERVTMNCKSSQSLLYSTNQKNYLAWYQQKPGQSPKLLIYWASTRESGVPDRFSGSGSGTDFTLTI -

Methadone Maintenance
Methadone Maintenance Judith Martin, MD Medical Director BAART Turk Street Clinic San Francisco, CA Dr. Martin, Disclosures • No conflict of interest to disclose. • No discussion of off-label use. • Special thanks to Dr. Thomas Payte for allowing me to use some of his great slides. Physicians Working in MMT Will Know: • How to safely induce patients to MMT. • How to adjust methadone dosing for maximum effectiveness. • How to detect and manage MMT side effects and medication interactions. • How to work within legal/regulatory framework of 42 CFR part 8. Safety Considerations in MMT: • Avoid sedation and respiratory depression (stay within tolerance) • Minimize side effects of constipation, sweating, hypogonadism • Alertness to potential medication interactions, QT/cardiac risk • Minimize diversion, accidental ingestion or dosing errors Safe Induction: “Start Low go Slow” • Methadone-related deaths during MMT occur during the first 10 days of treatment and are more common with higher induction doses. • There is no way of directly measuring tolerance to methadone. Estimate of opioid tolerance is based on history and physical, supported by toxicology tests. Methadone Deaths in Treatment • Induction risk 7 times greater than active heroin use. Methadone induces its own metabolic rate over time. Eventual dose needed will be usually much higher than initial tolerance. • Most deaths also show benzodiazepines on toxicology. This is true of most unintentional opiate poisonings. Methadone is an Unusual Opioid: • Slow onset of action: patient starts to ‘feel’ the swallowed dose 30-45 minutes later. • Delayed peak action: greatest effect from single dose is 2-4 hours post ingestion. • Tissue stores: methadone deposited in tissue over 3-7 days to reach steady state. -

Lanoxin, Tablet
NEW ZEALAND DATA SHEET 1. LANOXIN Digoxin tablets 62.5mcg (0.0625mg) and 250mcg (0.250mg); Paediatric Elixir 50mcg/mL 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Tablets: Each tablet contains 62.5mcg (0.0625mg), 250mcg (0.250mg) Digoxin BP Paediatric Elixir: 50mcg Digoxin BP in each 1mL For full list of excipients, see section 6.1 3. PHARMACEUTICAL FORM Tablets PG 62.5mcg Blue, round, biconvex tablets debossed “DO6” and each containing 62.5mcg (0.0625mg) Digoxin BP. Tablets 250mcg White, round, biconvex tablets, bisected and debossed ‘DO25” and each containing 250mcg (0.250mg) Digoxin BP. Paediatric Elixir 50mcg/mL A clear, yellow, lime flavoured solution containing 50mcg (0.050mg) Digoxin BP in each 1mL of sweetened, aqueous-alcoholic vehicle. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Cardiac Failure LANOXIN is indicated in the management of chronic cardiac failure where the dominant problem is systolic dysfunction. Its therapeutic benefit is greatest in those patients with ventricular dilatation. LANOXIN is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Supraventricular Arrhythmias LANOXIN is indicated in the management of certain supraventricular arrhythmias, particularly atrial flutter and fibrillation, where a major beneficial effect is reduction of the ventricular rate. 4.2 Dose and method of administration The dose of LANOXIN for each patient has to be tailored individually according to age, lean body weight and renal function. Suggested doses are intended only as an initial guide. LANOXIN PG Oral Solution, 50mcg in 1mL, is supplied with a graduated pipette and this should be used for measurement of all doses. Adults and children over 10 years Rapid Oral Loading 1 If medically appropriate, rapid digitalisation may be achieved in a number of ways, such as the following: 750 to 1500mcg (0.75 to 1.5mg) as a single dose.