1 Pharmacology Week 1 – General
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1: Clinical Pharmacokinetics 1
1: CLINICAL PHARMACOKINETICS 1 General overview: clinical pharmacokinetics, 2 Pharmacokinetics, 4 Drug clearance (CL), 6 Volume of distribution (Vd), 8 The half-life (t½), 10 Oral availability (F), 12 Protein binding (PB), 14 pH and pharmacokinetics, 16 1 Clinical pharmacokinetics General overview General overview: clinical pharmacokinetics 1 The ultimate aim of drug therapy is to achieve effi cacy without toxicity. This involves achieving a plasma concentration (Cp) within the ‘therapeutic window’, i.e. above the min- imal effective concentration (MEC), but below the minimal toxic concentration (MTC). Clinical pharmacokinetics is about all the factors that determine variability in the Cp and its time-course. The various factors are dealt with in subsequent chapters. Ideal therapeutics: effi cacy without toxicity Minimum Toxic Concentration (MTC) Ideal dosing Minimum Effective Concentration (MEC) Drug concentration Time The graph shows a continuous IV infusion at steady state, where the dose-rate is exactly appropriate for the patient’s clearance (CL). Inappropriate dosing Dosing too high in relation to the patient’s CL – toxicity likely Minimum Toxic Concentration (MTC) Minimum Effective Concentration (MEC) Dosing too low in relation to the Drug concentration patient’s CL – drug may be ineffective Time Some reasons for variation in CL Low CL High CL Normal variation Normal variation Renal impairment Increased renal blood fl ow Genetic poor metabolism Genetic hypermetabolism Liver impairment Enzyme induction Enzyme inhibition Old age/neonate 2 General overview Clinical Pharmacokinetics Pharmacokinetic factors determining ideal therapeutics If immediate effect is needed, a loading dose (LD) must be given to achieve a desired 1 concentration. The LD is determined by the volume of distribution (Vd). -

Principle of Pharmacodynamics
Principle of pharmacodynamics Dr. M. Emamghoreishi Full Professor Department of Pharmacology Medical School Shiraz University of Medical Sciences Email:[email protected] Reference: Basic & Clinical Pharmacology: Bertrum G. Katzung and Anthony J. Treveror, 13th edition, 2015, chapter 20, p. 336-351 Learning Objectives: At the end of sessions, students should be able to: 1. Define pharmacology and explain its importance for a clinician. 2. Define ―drug receptor‖. 3. Explain the nature of drug receptors. 4. Describe other sites of drug actions. 5. Explain the drug-receptor interaction. 6. Define the terms ―affinity‖, ―intrinsic activity‖ and ―Kd‖. 7. Explain the terms ―agonist‖ and ―antagonist‖ and their different types. 8. Explain chemical and physiological antagonists. 9. Explain the differences in drug responsiveness. 10. Explain tolerance, tachyphylaxis, and overshoot. 11. Define different dose-response curves. 12. Explain the information that can be obtained from a graded dose-response curve. 13. Describe the potency and efficacy of drugs. 14. Explain shift of dose-response curves in the presence of competitive and irreversible antagonists and its importance in clinical application of antagonists. 15. Explain the information that can be obtained from a quantal dose-response curve. 16. Define the terms ED50, TD50, LD50, therapeutic index and certain safety factor. What is Pharmacology?It is defined as the study of drugs (substances used to prevent, diagnose, and treat disease). Pharmacology is the science that deals with the interactions betweena drug and the bodyor living systems. The interactions between a drug and the body are conveniently divided into two classes. The actions of the drug on the body are termed pharmacodynamicprocesses.These properties determine the group in which the drug is classified, and they play the major role in deciding whether that group is appropriate therapy for a particular symptom or disease. -

Opioid Receptorsreceptors
OPIOIDOPIOID RECEPTORSRECEPTORS defined or “classical” types of opioid receptor µ,dk and . Alistair Corbett, Sandy McKnight and Graeme Genes encoding for these receptors have been cloned.5, Henderson 6,7,8 More recently, cDNA encoding an “orphan” receptor Dr Alistair Corbett is Lecturer in the School of was identified which has a high degree of homology to Biological and Biomedical Sciences, Glasgow the “classical” opioid receptors; on structural grounds Caledonian University, Cowcaddens Road, this receptor is an opioid receptor and has been named Glasgow G4 0BA, UK. ORL (opioid receptor-like).9 As would be predicted from 1 Dr Sandy McKnight is Associate Director, Parke- their known abilities to couple through pertussis toxin- Davis Neuroscience Research Centre, sensitive G-proteins, all of the cloned opioid receptors Cambridge University Forvie Site, Robinson possess the same general structure of an extracellular Way, Cambridge CB2 2QB, UK. N-terminal region, seven transmembrane domains and Professor Graeme Henderson is Professor of intracellular C-terminal tail structure. There is Pharmacology and Head of Department, pharmacological evidence for subtypes of each Department of Pharmacology, School of Medical receptor and other types of novel, less well- Sciences, University of Bristol, University Walk, characterised opioid receptors,eliz , , , , have also been Bristol BS8 1TD, UK. postulated. Thes -receptor, however, is no longer regarded as an opioid receptor. Introduction Receptor Subtypes Preparations of the opium poppy papaver somniferum m-Receptor subtypes have been used for many hundreds of years to relieve The MOR-1 gene, encoding for one form of them - pain. In 1803, Sertürner isolated a crystalline sample of receptor, shows approximately 50-70% homology to the main constituent alkaloid, morphine, which was later shown to be almost entirely responsible for the the genes encoding for thedk -(DOR-1), -(KOR-1) and orphan (ORL ) receptors. -

Prof. Hanan Hagar Quantitative Aspects of Drugs
Quantitative aspects of drugs Prof. hanan Hagar Ilos Determine quantitative aspects of drug receptor binding. Recognize concentration binding curves. Identify dose response curves and the therapeutic utility of these curves. Classify different types of antagonism QUANTIFY ASPECTS OF DRUG ACTION Bind Initiate Occupy Activate D + R D R DR* RESPONSE[R] Relate concentration [C] of D used (x- axis) Relate concentration [C] of D used (x- to the binding capacity at receptors (y-axis) axis) to response produced (y-axis) Concentration-Binding Curve Dose Response Curves AFFINITY EFFICACY POTENCY Concentration binding curves Is a correlation between drug concentration [C] used (x- axis) and drug binding capacity at receptors [B] (y-axis). i.e. relation between concentration & drug binding Concentration-Binding curves are used to determine: oBmax (the binding capacity) is the total density of receptors in the tissues. oKD50 is the concentration of drug required to occupy 50% of receptors at equilibrium. oThe affinity of drug for receptor The higher the affinity of D for receptor the lower is the KD i.e. inverse relation Concentration-Binding Curve (Bmax): Total density of receptors in the tissue K D KD (kD )= [C] of D required to occupy 50% of receptors at equilibrium Dose -response curves o Used to study how response varies with the concentration or dose. o Is a correlation between drug concentration [D] used (x- axis) and drug response [R] (y-axis). o i.e. relation between concentration & Response TYPES of Dose -response curves Graded dose-response curve Quantal dose-response curve (all or none). Graded Dose-response Curve o Response is gradual o Gradual increase in response by increasing the dose (continuous response). -

Pharmacodynamics Drug Receptor Interactions Part-2
Pharmacodynamics: (Drug Receptor Interactions, Part 2) ………………………………………………………………………………………………………………………………………………………………………………………………………………… VPT: Unit I; Lecture-22 (Dated 03.12.2020) Dr. Nirbhay Kumar Asstt. Professor & Head Deptt. of Veterinary Pharmacology & Toxicology Bihar Veterinary College, Bihar Animal Sciences University, Patna Drug Receptor Interactions Agonist It is a drug that possesses affinity for a particular receptor and causes a change in the receptor that result in an observable effect. Full agonist: Produces a maximal response by occupying all or a fraction of receptors. (Affinity=1, Efficacy=1) Partial agonist: Produces less than a maximal response even when the drug occupies all of the receptors. (Affinity=1, Efficacy= 0 to 1) Inverse agonist: Activates a receptor to produce an effect in the opposite direction to that of the well recognized agonist. (Affinity=1, Efficacy= –1 to 0). Source: Rang & Dale’s Pharmacology, Elsevier Source: Good & Gilman’s The Pharmacological Basis of Therapeutics, 13th Edn. Antagonist An antagonist is a drug that blocks the response produced by an agonist. Antagonists interact with the receptor or other components of the effector mechanism, but antagonists are devoid of intrinsic activity (Affinity=1, Efficacy=0). Antagonist contd… Competitive Antagonism: It is completely reversible; an increase in the concentration of the agonist in the bio-phase will overcome the effect of the antagonist. Example: Atropine (Antimuscarinic agent) Diphenhydramine (H1 receptor blocker) Non-competitive antagonism: The agonist has no influence upon the degree of antagonism or its reversibility. Example: Platelet inhibiting action of aspirin (The thromboxane synthase enzyme of platelets is irreversibly inhibited by aspirin, a process that is reversed only by production of new platelets). -

CPIC Guideline for Pharmacogenetics-Guided Warfarin Dosing – Supplement V2.0 1 Table of Contents Guideline Updates
Supplement to: Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for Pharmacogenetics-guided Warfarin Dosing: 2016 Update Julie A. Johnson1, Kelly Caudle2, Li Gong3, Michelle Whirl-Carrillo3, C. Michael Stein4, Stuart A. Scott5, Ming Ta Michael Lee6 , Brian F. Gage7, Stephen E. Kimmel8,9, Minoli A. Perera10, Jeffrey L. Anderson11, Munir Pirmohamed12, Teri E. Klein3, Nita A. Limdi13, Larisa H. Cavallari1, Mia Wadelius14 1Department of Pharmacotherapy and Translational Research, College of Pharmacy, and Center for Pharmacogenomics, University of Florida, Gainesville, Florida, USA 2Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN 3Department of Genetics, Stanford University, Stanford, California, USA 4Division of Clinical Pharmacology Vanderbilt Medical School, Nashville, TN, USA 5Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA 6Laboratory for International Alliance on Genomic Research, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan; National Center for Genome Medicine; Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan; Genomic Medicine Institute Geisinger Health system, Danville, PA 7Department of Internal Medicine, Washington University in St. Louis, St. Louis, Missouri 8Center for Clinical Epidemiology and Biostatistics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA 9Department of Medicine and Department of Biostatistics and Epidemiology, University of Pennsylvania -

Measuring Ligand Efficacy at the Mu- Opioid Receptor Using A
RESEARCH ARTICLE Measuring ligand efficacy at the mu- opioid receptor using a conformational biosensor Kathryn E Livingston1,2, Jacob P Mahoney1,2, Aashish Manglik3, Roger K Sunahara4, John R Traynor1,2* 1Department of Pharmacology, University of Michigan Medical School, Ann Arbor, United States; 2Edward F Domino Research Center, University of Michigan, Ann Arbor, United States; 3Department of Pharmaceutical Chemistry, School of Pharmacy, University of California San Francisco, San Francisco, United States; 4Department of Pharmacology, University of California San Diego School of Medicine, La Jolla, United States Abstract The intrinsic efficacy of orthosteric ligands acting at G-protein-coupled receptors (GPCRs) reflects their ability to stabilize active receptor states (R*) and is a major determinant of their physiological effects. Here, we present a direct way to quantify the efficacy of ligands by measuring the binding of a R*-specific biosensor to purified receptor employing interferometry. As an example, we use the mu-opioid receptor (m-OR), a prototypic class A GPCR, and its active state sensor, nanobody-39 (Nb39). We demonstrate that ligands vary in their ability to recruit Nb39 to m- OR and describe methadone, loperamide, and PZM21 as ligands that support unique R* conformation(s) of m-OR. We further show that positive allosteric modulators of m-OR promote formation of R* in addition to enhancing promotion by orthosteric agonists. Finally, we demonstrate that the technique can be utilized with heterotrimeric G protein. The method is cell- free, signal transduction-independent and is generally applicable to GPCRs. DOI: https://doi.org/10.7554/eLife.32499.001 *For correspondence: [email protected] Competing interests: The authors declare that no Introduction competing interests exist. -

Efavirenz) Capsules and Tablets 3 Rx Only
1 SUSTIVA® 2 (efavirenz) capsules and tablets 3 Rx only 4 DESCRIPTION 5 SUSTIVA® (efavirenz) is a human immunodeficiency virus type 1 (HIV-1) specific, non- 6 nucleoside, reverse transcriptase inhibitor (NNRTI). 7 Capsules: SUSTIVA is available as capsules for oral administration containing either 8 50 mg, 100 mg, or 200 mg of efavirenz and the following inactive ingredients: lactose 9 monohydrate, magnesium stearate, sodium lauryl sulfate, and sodium starch glycolate. 10 The capsule shell contains the following inactive ingredients and dyes: gelatin, sodium 11 lauryl sulfate, titanium dioxide, and/or yellow iron oxide. The capsule shells may also 12 contain silicon dioxide. The capsules are printed with ink containing carmine 40 blue, 13 FD&C Blue No. 2, and titanium dioxide. 14 Tablets: SUSTIVA is available as film-coated tablets for oral administration containing 15 600 mg of efavirenz and the following inactive ingredients: croscarmellose sodium, 16 hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, microcrystalline 17 cellulose, and sodium lauryl sulfate. The film coating contains Opadry® Yellow and 18 Opadry® Clear. The tablets are polished with carnauba wax and printed with purple ink, 19 Opacode® WB. 20 Efavirenz is chemically described as (S)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4- 21 (trifluoromethyl)-2H-3,1-benzoxazin-2-one. 22 Its empirical formula is C14H9ClF3NO2 and its structural formula is: 1 of 45 Approved v2.0 F C 3 Cl O NO 23 H 24 Efavirenz is a white to slightly pink crystalline powder with a molecular mass of 315.68. 25 It is practically insoluble in water (<10 µg/mL). -

Factors Affecting Drug Response2
Types of drug concentration– response relationship A. Graded drug concentration-Response Relationships As the dose of a drug is increased, the response (effect) of the tissue or organ is also increased. The efficacy (Emax) and potency (ED50) parameters are derived from these data. 112 GRADED DOSE- RESPONSE CURVE QUANTAL DOSE- RESPONSE CURVE 113 B. Quantal or all or none dose response relation ship. The Plot of the fraction of the population that responds at each dose of the drug versus the log of the dose administered. responses follow all or none phenomenon – that means the individual of the responding system either respond to their maximum limit or not at all to a dose of drug and there is no gradation of response. population studies. relates dose to frequency of effect . The median effective (ED50), median toxic (TD50) ,and median lethal doses (LD50) are extracted from experiments carried out in this manner. 114 Median effective dose (ED50):the dose at which 50% of individuals exhibit the specified quantal effect. Median toxic dose (TD50) :the dose required to produce a particular toxic effect in 50% of animals. Median lethal dose (LD50): is the lethal dose that causes death in 50% animal under experiment. 115 119 1. Excretion - Alteration of urine PH. e.g. Phanobarbitone + NaHCo3 - Alteration of active tubular secretion e.g. Probenecid + peincillin. II – Pharmcodynamic interaction - It occurs by modification of pharmacological response of one drug by another without altering the concentration of the drug in the tissue or tissue fluid. 1. Additive – Occurs when the combined effect of two drugs is equal to the sum of the effects of each agent given alone. -

The Importance of Serotonergic and Adrenergic Receptors for the Induction and Expression of One-Trial Cocaine-Induced Behavioral Sensitization" (2016)
California State University, San Bernardino CSUSB ScholarWorks Electronic Theses, Projects, and Dissertations Office of aduateGr Studies 12-2016 THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE- TRIAL COCAINE-INDUCED BEHAVIORAL SENSITIZATION Krista N. Rudberg California State University - San Bernardino Follow this and additional works at: https://scholarworks.lib.csusb.edu/etd Part of the Biological Psychology Commons, and the Pharmacology Commons Recommended Citation Rudberg, Krista N., "THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE-TRIAL COCAINE-INDUCED BEHAVIORAL SENSITIZATION" (2016). Electronic Theses, Projects, and Dissertations. 420. https://scholarworks.lib.csusb.edu/etd/420 This Thesis is brought to you for free and open access by the Office of aduateGr Studies at CSUSB ScholarWorks. It has been accepted for inclusion in Electronic Theses, Projects, and Dissertations by an authorized administrator of CSUSB ScholarWorks. For more information, please contact [email protected]. THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE-TRIAL COCAINE- INDUCED BEHAVIORAL SENSITIZATION A Thesis Presented to the Faculty of California State University, San Bernardino In Partial Fulfillment of the Requirements for the Degree Master of Arts in General/Experimental Psychology by Krista Nicole Rudberg December 2016 THE IMPORTANCE OF SEROTONERGIC AND ADRENERGIC RECEPTORS FOR THE INDUCTION AND EXPRESSION OF ONE-TRIAL COCAINE- INDUCED BEHAVIORAL SENSITIZATION A Thesis Presented to the Faculty of California State University, San Bernardino by Krista Nicole Rudberg December 2016 Approved by: Sanders McDougall, Committee Chair, Psychology Cynthia Crawford, Committee Member Matthew Riggs, Committee Member © 2016 Krista Nicole Rudberg ABSTRACT Addiction is a complex process in which behavioral sensitization may be an important component. -
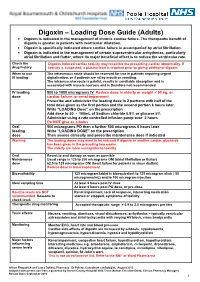
Digoxin – Loading Dose Guide (Adults) Digoxin Is Indicated in the Management of Chronic Cardiac Failure
Digoxin – Loading Dose Guide (Adults) Digoxin is indicated in the management of chronic cardiac failure. The therapeutic benefit of digoxin is greater in patients with ventricular dilatation. Digoxin is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Digoxin is indicated in the management of certain supraventricular arrhythmias, particularly atrial fibrillation and flutter, where its major beneficial effect is to reduce the ventricular rate. Check the Digoxin-induced cardiac toxicity may resemble the presenting cardiac abnormality. If drug history toxicity is suspected, a plasma level is required prior to giving additional digoxin. When to use The intravenous route should be reserved for use in patients requiring urgent IV loading digitalisation, or if patients are nil by mouth or vomiting. The intramuscular route is painful, results in unreliable absorption and is associated with muscle necrosis and is therefore not recommended. IV loading 500 to 1000 micrograms IV Reduce dose in elderly or weight < 50 kg, or dose cardiac failure, or renal impairment Prescribe and administer the loading dose in 2 portions with half of the total dose given as the first portion and the second portion 6 hours later. Write “LOADING Dose” on the prescription Add dose to 50 - 100mL of Sodium chloride 0.9% or glucose 5% Administer using a rate controlled infusion pump over 2 hours Do NOT give as a bolus Oral 500 micrograms PO then a further 500 micrograms 6 hours later loading Write “LOADING DOSE” on the prescription dose Then assess clinically and prescribe maintenance dose if indicated Warning The loading doses may need to be reduced if digoxin or another cardiac glycoside has been given in the preceding two weeks. -
![Response Vs. Log [L] - Full Agonist](https://docslib.b-cdn.net/cover/7019/response-vs-log-l-full-agonist-937019.webp)
Response Vs. Log [L] - Full Agonist
DavidsonX – D001x – Medicinal Chemistry Chapter 5 – Receptors Part 2 – Ligands Video Clip – Ligand Types Ligands can have different effects on a receptor. Each type of ligand can be readily classified according to its behavior. A type of ligand is the full agonist. The term agonist refers to a compound that binds a receptor and elicits a response (E). Full agonists elicit the same level of full response (E = Emax = 100%) as the endogenous ligand of the receptor. Graphically, a receptor-ligand interaction is plotted as response (E/Emax) vs. log [L]. The relationship is sigmoidal. A full agonist approaches full response (E/Emax = 1.0) as log [L] reaches relatively high levels. response vs. log [L] - full agonist 1 0.9 0.8 0.7 x a 0.6 m E 0.5 / E 0.4 0.3 0.2 0.1 0 log [L] Two ligands can achieve a full response without being equivalent. Ligands can differ with respect to the concentration required to trigger a response. A ligand that affects a response at a lower concentration has a higher potency. Potencies are measured as the effective ligand concentration required to reach a 50% response – EC50 or, in these graphs, log [EC50]. A more potent ligand has a lower EC50 value. full agonist comparison 1 0.9 full agonist 1 0.8 (more potent) full agonist 2 0.7 (less potent) x a 0.6 log EC m 50 E 0.5 / E 0.4 0.3 log EC 0.2 50 0.1 0 log [L] Partial agonists also cause a response, but they cannot reach the same, 100% response level of the endogenous ligand.