Pharmacodynamics Drug Receptor Interactions Part-2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacodynamics - I
Pharmacodynamics - I Dr. Jyoti M. Benni Dept. of Pharmacology USM-KLE, IMP Belgaum Learning outcomes • Describe the principles of pharmacodynamics with regard to the potential targets of -drug action -receptor types -dose-response relationship (curve) -therapeutic index 2 Introduction: PK & PD 3 Pharmacodynamics Pharmacodynamics is the study of actions of the drug on the body and their mechanism of action. Stimulation Depression Irritation Replacement Modify immune status Anti-infective / Cytotoxic action 4 Mechanisms of Drug Action Non-receptor mediated Receptor mediated • Physical • Receptors on the cell • Chemical membrane • Enzymes • Ion channels • Transporters • Receptors inside the cell • Antibody • Placebo 5 Non – receptor mediated mechanisms… Physical property . Physical property of the drug is responsible E.g. Adsorption: activated charcoal in treatment of poisoning Osmotic activity: magnesium sulfate for constipation Radioactivity: radioactive iodine (I131 ) for hyperthyroidism Radioopacity: barium sulfate as contrast media 6 Non – receptor mediated mechanisms… Chemical action Antacids - neutralize gastric acid Chelating agents (EDTA) Used in heavy metal (LEAD)poisoning treatment Oxidizing agents potassium permanganate as germicidal agent 7 Non – receptor mediated mechanisms… Enzymes as targets of drug action Enzymes Inhibition Stimulation Enzyme Nonspecific Specific induction Competitive Noncompetitive 8 Non – receptor mediated mechanisms… Enzyme stimulation: • Reactivation e.g. Injection pralidoxime → for treatment of Organophosphorus -

Activity Intrinsic
Vol. (Suppl. ) Intrinsic 201 Activity www.IntrinsicActivity.org Published by th ISSN 2309-8503 Austrian Pharmacological Society Dopamine 2016 Vienna, 5–8 September 2016 MAEETING BSTRACTS Intrinsic Activity is an online, open-access publication medium published by the Austrian Pharmacological Society (APHAR). The Journal welcomes contributions in the fields of Pharmacology, Pharmacotherapy and other fields in biomedicine. Contributions may be of type meeting abstracts, research articles, position papers, commentaries or similar. For submission instructions and all other information regarding publication in the journal visit: www.IntrinsicActivity.org Correspondence Intrinsic Activity c/o Institute for Experimental and Clinical Pharmacology Medical University of Graz Universitätsplatz 4 8010 Graz, Austria Tel.: +43 (316) 380-4305 Fax: +43 (316) 380-9645 E-mail: [email protected] Website: www.IntrinsicActivity.org ISSN: 2309-8503 Austrian Pharmacological Society c/o Institute of Pharmacology Centre for Physiology and Pharmacology Medical University of Vienna Währinger Straße 13a 1090 Wien, Austria E-mail: [email protected] Copyright, open access and permission to use Articles are published under a Creative Commons license (Creative Commons, attribution, non-commercial), that allows reuse subject only to the use being non-commercial and the article being fully attributed. The Publisher and Austrian Pharmacological Society retain the license that allows publishing of the articles in Intrinsic Activity, any derivative product or any other Intrinsic Activity product (present or future) and allows sub-licensing such rights and exploit all subsidiary rights. Authors retain the license to use their articles for their own non-commercial purposes, specifically: Posting a pdf of their own article on their own personal or institutional website for which no charge for access is made. -

Principle of Pharmacodynamics
Principle of pharmacodynamics Dr. M. Emamghoreishi Full Professor Department of Pharmacology Medical School Shiraz University of Medical Sciences Email:[email protected] Reference: Basic & Clinical Pharmacology: Bertrum G. Katzung and Anthony J. Treveror, 13th edition, 2015, chapter 20, p. 336-351 Learning Objectives: At the end of sessions, students should be able to: 1. Define pharmacology and explain its importance for a clinician. 2. Define ―drug receptor‖. 3. Explain the nature of drug receptors. 4. Describe other sites of drug actions. 5. Explain the drug-receptor interaction. 6. Define the terms ―affinity‖, ―intrinsic activity‖ and ―Kd‖. 7. Explain the terms ―agonist‖ and ―antagonist‖ and their different types. 8. Explain chemical and physiological antagonists. 9. Explain the differences in drug responsiveness. 10. Explain tolerance, tachyphylaxis, and overshoot. 11. Define different dose-response curves. 12. Explain the information that can be obtained from a graded dose-response curve. 13. Describe the potency and efficacy of drugs. 14. Explain shift of dose-response curves in the presence of competitive and irreversible antagonists and its importance in clinical application of antagonists. 15. Explain the information that can be obtained from a quantal dose-response curve. 16. Define the terms ED50, TD50, LD50, therapeutic index and certain safety factor. What is Pharmacology?It is defined as the study of drugs (substances used to prevent, diagnose, and treat disease). Pharmacology is the science that deals with the interactions betweena drug and the bodyor living systems. The interactions between a drug and the body are conveniently divided into two classes. The actions of the drug on the body are termed pharmacodynamicprocesses.These properties determine the group in which the drug is classified, and they play the major role in deciding whether that group is appropriate therapy for a particular symptom or disease. -

Measuring Ligand Efficacy at the Mu- Opioid Receptor Using A
RESEARCH ARTICLE Measuring ligand efficacy at the mu- opioid receptor using a conformational biosensor Kathryn E Livingston1,2, Jacob P Mahoney1,2, Aashish Manglik3, Roger K Sunahara4, John R Traynor1,2* 1Department of Pharmacology, University of Michigan Medical School, Ann Arbor, United States; 2Edward F Domino Research Center, University of Michigan, Ann Arbor, United States; 3Department of Pharmaceutical Chemistry, School of Pharmacy, University of California San Francisco, San Francisco, United States; 4Department of Pharmacology, University of California San Diego School of Medicine, La Jolla, United States Abstract The intrinsic efficacy of orthosteric ligands acting at G-protein-coupled receptors (GPCRs) reflects their ability to stabilize active receptor states (R*) and is a major determinant of their physiological effects. Here, we present a direct way to quantify the efficacy of ligands by measuring the binding of a R*-specific biosensor to purified receptor employing interferometry. As an example, we use the mu-opioid receptor (m-OR), a prototypic class A GPCR, and its active state sensor, nanobody-39 (Nb39). We demonstrate that ligands vary in their ability to recruit Nb39 to m- OR and describe methadone, loperamide, and PZM21 as ligands that support unique R* conformation(s) of m-OR. We further show that positive allosteric modulators of m-OR promote formation of R* in addition to enhancing promotion by orthosteric agonists. Finally, we demonstrate that the technique can be utilized with heterotrimeric G protein. The method is cell- free, signal transduction-independent and is generally applicable to GPCRs. DOI: https://doi.org/10.7554/eLife.32499.001 *For correspondence: [email protected] Competing interests: The authors declare that no Introduction competing interests exist. -

Pharmacogenomic and Structural Analysis of Constitutive G Protein–Coupled Receptor Activity
ANRV298-PA47-02 ARI 4 December 2006 20:18 Pharmacogenomic and Structural Analysis of Constitutive G Protein–Coupled Receptor Activity Martine J. Smit,1 Henry F. Vischer,1 Remko A. Bakker,1 Aldo Jongejan,1 Henk Timmerman,1 Leonardo Pardo,2 and Rob Leurs1 1Leiden/Amsterdam Center for Drug Research, Division of Medicinal Chemistry, Vrije Universiteit, Faculty of Sciences, Department of Chemistry, 1081 HV Amsterdam, The Netherlands; email: [email protected] 2Laboratorio de Medicina Computacional, Unidad de Bioestadistica, Facultad de Medicina, Universidad Autonoma de Barcelona, Barcelona, Spain Annu. Rev. Pharmacol. Toxicol. 2007. 47:53–87 Key Words First published online as a Review in Advance on constitutive activity; inverse agonism, receptor structure, receptor October 9, 2006 motifs The Annual Review of Pharmacology and Toxicology is online at http://pharmtox.annualreviews.org Abstract This article’s doi: G protein–coupled receptors (GPCRs) respond to a chemically di- 10.1146/annurev.pharmtox.47.120505.105126 verse plethora of signal transduction molecules. The notion that Copyright c 2007 by Annual Reviews. GPCRs also signal without an external chemical trigger, i.e., in a All rights reserved constitutive or spontaneous manner, resulted in a paradigm shift by Universitat Autonoma de Barcelona on 01/09/07. For personal use only. 0362-1642/07/0210-0053$20.00 in the field of GPCR pharmacology. The discovery of constitutive GPCR activity and the fact that GPCR binding and signaling can be strongly affected by a single point mutation drew attention to Annu. Rev. Pharmacol. Toxicol. 2007.47:53-87. Downloaded from arjournals.annualreviews.org the evolving area of GPCR pharmacogenomics. -
![Response Vs. Log [L] - Full Agonist](https://docslib.b-cdn.net/cover/7019/response-vs-log-l-full-agonist-937019.webp)
Response Vs. Log [L] - Full Agonist
DavidsonX – D001x – Medicinal Chemistry Chapter 5 – Receptors Part 2 – Ligands Video Clip – Ligand Types Ligands can have different effects on a receptor. Each type of ligand can be readily classified according to its behavior. A type of ligand is the full agonist. The term agonist refers to a compound that binds a receptor and elicits a response (E). Full agonists elicit the same level of full response (E = Emax = 100%) as the endogenous ligand of the receptor. Graphically, a receptor-ligand interaction is plotted as response (E/Emax) vs. log [L]. The relationship is sigmoidal. A full agonist approaches full response (E/Emax = 1.0) as log [L] reaches relatively high levels. response vs. log [L] - full agonist 1 0.9 0.8 0.7 x a 0.6 m E 0.5 / E 0.4 0.3 0.2 0.1 0 log [L] Two ligands can achieve a full response without being equivalent. Ligands can differ with respect to the concentration required to trigger a response. A ligand that affects a response at a lower concentration has a higher potency. Potencies are measured as the effective ligand concentration required to reach a 50% response – EC50 or, in these graphs, log [EC50]. A more potent ligand has a lower EC50 value. full agonist comparison 1 0.9 full agonist 1 0.8 (more potent) full agonist 2 0.7 (less potent) x a 0.6 log EC m 50 E 0.5 / E 0.4 0.3 log EC 0.2 50 0.1 0 log [L] Partial agonists also cause a response, but they cannot reach the same, 100% response level of the endogenous ligand. -

Different Inverse Agonist Activities of P»-Adrenergic Receptor Antagonists—Pharmacological Characterization and Therapeutical
International Congress Series 1249 (2003) 39-53 Different inverse agonist activities of p»-adrenergic receptor antagonists—pharmacological characterization and therapeutical implications in the treatment of chronic heart failure Christoph Maack*, Michael Bòhm Medizinische Klinik und Poliklinik fiir Innere Medizin III, Universitat des Saarlandes, 66421 Homburg/Saai; Germany Received 16 April 2003; accepted 16 April 2003 Abstract The treatment of chronic heart failure with most p-adrenergic receptor (p-AR) antagonists leads to an improvement of symptoms and left ventricular function. However, only metoprolol, bisoprolol and carvedilol have been shown to reduce mortality in these patients. Bucindolol did not reduce mortality and xamoterol even increased it. These differences may be related to different inverse agonist or partial agonist activity of p-AR antagonists. This review focusses on the determination of different intrinsic activity of the mentioned p-AR antagonists in the human myocardium. Furthermore, the clinical impact of these differences is examined. In this regard, the effect of the different p-AR antagonists on p-AR regulation, minimum heart rate and exercise tolerance, as well as prognosis, is highlighted. It is concluded that the degree of inverse agonism of a p-AR antagonist determines the degree of p-AR resensitization, reduction of minimum heart rate, improvement of exercise tolerance and possibly also improvement of prognosis of patients with chronic heart failure. © 2003 Elsevier Science B.V. All rights reserved. Key\vords: Inverse agonism; p-adrenergic receptors; p-blockers, Heart faitee * Corresponding author. Current address: The Johns Hopkins University, Institute of Molecular Cardio- biology, Division of Cardiology, 720 Rutland Ave., 844 Ross Bldg., Baltimore, MD 21205-2195, USA. -

Anew Drug Design Strategy in the Liht of Molecular Hybridization Concept
www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 “Drug Design strategy and chemical process maximization in the light of Molecular Hybridization Concept.” Subhasis Basu, Ph D Registration No: VB 1198 of 2018-2019. Department Of Chemistry, Visva-Bharati University A Draft Thesis is submitted for the partial fulfilment of PhD in Chemistry Thesis/Degree proceeding. DECLARATION I Certify that a. The Work contained in this thesis is original and has been done by me under the guidance of my supervisor. b. The work has not been submitted to any other Institute for any degree or diploma. c. I have followed the guidelines provided by the Institute in preparing the thesis. d. I have conformed to the norms and guidelines given in the Ethical Code of Conduct of the Institute. e. Whenever I have used materials (data, theoretical analysis, figures and text) from other sources, I have given due credit to them by citing them in the text of the thesis and giving their details in the references. Further, I have taken permission from the copyright owners of the sources, whenever necessary. IJCRT2012039 International Journal of Creative Research Thoughts (IJCRT) www.ijcrt.org 284 www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 f. Whenever I have quoted written materials from other sources I have put them under quotation marks and given due credit to the sources by citing them and giving required details in the references. (Subhasis Basu) ACKNOWLEDGEMENT This preface is to extend an appreciation to all those individuals who with their generous co- operation guided us in every aspect to make this design and drawing successful. -

The Pharmacological Studies on Intrinsic Activities of Acetylcholine, Methacholine, and Carbachol (Homologous Drugs) on Isolated Rat Ileum Preparation
Journal of Medicine and Medical Science Vol. 2(8) pp. 1047-1049, August 2011 Available online@ http://www.interesjournals.org/JMMS Copyright © 2011 International Research Journals Short Communication The pharmacological studies on intrinsic activities of acetylcholine, methacholine, and carbachol (Homologous Drugs) on isolated rat ileum preparation Peter I. Aziba* 1, Sokan J.O. 2, Ifedayo O. 2 and Kasim L.S. 2 1*Department of Pharmacology and 2Department of Pharmaceutical Chemistry Olabisi Onabanjo university, Oachs, Ago_Iwoye. Accepted 24 August, 2011 This study investigated the intrinsic activities of equimolar concentrations of three homologous drugs Actylcholine (Ach), Methacholine (Mch) and Carbachol (Cch) on isolated rat ileum preparation. Contractions were monitored on smoked drum fixed on a kymograph. Heights of contractions were measured in (mm) in order to assess their efficacies.In some experiments Ach- induced contractions were measured in the presence of atropine a competitive muscarinic antagonist. The result showed order of potencies, ranked as Cch>Mch>Ach. This result may explain the low intrinsic activities of Ach when compared to Cch, Mch, due to effect of acetylcholinesterase an enzyme which hydrolyze Ach and limit its efficacy in therapeutic use. Keywords: Intrinsic, homologous drugs, rat ileum, efficacy. INTRODUCTION Theoretically, the term intrinsic activity of a drug was first MATERIALS AND METHODS introduced by Ariens and Van Rossum (1957). The term has been used a great deal in their attempt to explain Rats between (200-305g) were used. The animals were why all drugs do not act in accordance with Clark’s theory maintained in well ventilated conditions, under constant of drug receptor interaction. -

Volume C, Module 2 Opioids: Basics of Addiction; Treatment with Agonists, Partial Agonists, and Antagonists
VolumeVolume C,C, ModuleModule 22 Opioids:Opioids: BasicsBasics ofof Addiction;Addiction; TreatmentTreatment withwith Agonists,Agonists, PartialPartial Agonists,Agonists, andand AntagonistsAntagonists Treatnet Training Volume C: Module 2 – Updated 18 October 2007 ModuleModule 2:2: TrainingTraining goalsgoals ToTo describedescribe the:the: ¾ KeyKey componentscomponents ofof opiateopiate addictionaddiction andand itsits medicalmedical // psychiatricpsychiatric consequencesconsequences ¾ BenefitsBenefits andand limitationslimitations ofof methadonemethadone asas aa pharmacotherapypharmacotherapy forfor opiateopiate dependencedependence ¾ BenefitsBenefits andand limitationslimitations ofof buprenorphinebuprenorphine asas aa pharmacotherapypharmacotherapy forfor opiateopiate dependencedependence ¾ BenefitsBenefits andand limitationslimitations ofof narcoticnarcotic antagonistsantagonists forfor overdoseoverdose (naloxone)(naloxone) andand relapserelapse preventionprevention (naltrexone)(naltrexone) forfor opiateopiate dependencedependence ModuleModule 2:2: WorkshopsWorkshops WorkshopWorkshop 1:1: Opiates:Opiates: WhatWhat theythey are,are, problemsproblems associatedassociated withwith theirtheir use,use, andand medicalmedical treatmenttreatment implicationsimplications WorkshopWorkshop 2:2: OpiateOpiate addictionaddiction treatmenttreatment withwith methadonemethadone WorkshopWorkshop 3:3: OpiateOpiate addictionaddiction treatmenttreatment withwith buprenorphinebuprenorphine WorkshopWorkshop 4:4: OpiateOpiate AntagonistAntagonist Treatment:Treatment: -

International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology
0031-6997/03/5504-597–606$7.00 PHARMACOLOGICAL REVIEWS Vol. 55, No. 4 Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 30404/1114803 Pharmacol Rev 55:597–606, 2003 Printed in U.S.A International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology RICHARD R. NEUBIG, MICHAEL SPEDDING, TERRY KENAKIN, AND ARTHUR CHRISTOPOULOS Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (R.R.N.); Institute de Recherches Internationales Servier, Neuilly sur Seine, France (M.S.); Systems Research, GlaxoSmithKline Research and Development, Research Triangle Park, North Carolina (T.K.); and Department of Pharmacology, University of Melbourne, Parkville, Australia (A.C.) Abstract ............................................................................... 597 I. Introduction............................................................................ 597 II. Working definition of a receptor .......................................................... 598 III. Use of drugs in definition of receptors or of signaling pathways ............................. 598 A. The expression of amount of drug: concentration and dose ............................... 598 Downloaded from 1. Concentration..................................................................... 598 2. Dose. ............................................................................ 598 B. General terms used to describe drug action ........................................... -
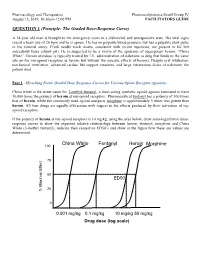
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined.