Cariprazine, a Dopamine D2/D3 Receptor Partial Agonist, Modulates ABCG2-Mediated Multidrug Resistance in Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Principle of Pharmacodynamics
Principle of pharmacodynamics Dr. M. Emamghoreishi Full Professor Department of Pharmacology Medical School Shiraz University of Medical Sciences Email:[email protected] Reference: Basic & Clinical Pharmacology: Bertrum G. Katzung and Anthony J. Treveror, 13th edition, 2015, chapter 20, p. 336-351 Learning Objectives: At the end of sessions, students should be able to: 1. Define pharmacology and explain its importance for a clinician. 2. Define ―drug receptor‖. 3. Explain the nature of drug receptors. 4. Describe other sites of drug actions. 5. Explain the drug-receptor interaction. 6. Define the terms ―affinity‖, ―intrinsic activity‖ and ―Kd‖. 7. Explain the terms ―agonist‖ and ―antagonist‖ and their different types. 8. Explain chemical and physiological antagonists. 9. Explain the differences in drug responsiveness. 10. Explain tolerance, tachyphylaxis, and overshoot. 11. Define different dose-response curves. 12. Explain the information that can be obtained from a graded dose-response curve. 13. Describe the potency and efficacy of drugs. 14. Explain shift of dose-response curves in the presence of competitive and irreversible antagonists and its importance in clinical application of antagonists. 15. Explain the information that can be obtained from a quantal dose-response curve. 16. Define the terms ED50, TD50, LD50, therapeutic index and certain safety factor. What is Pharmacology?It is defined as the study of drugs (substances used to prevent, diagnose, and treat disease). Pharmacology is the science that deals with the interactions betweena drug and the bodyor living systems. The interactions between a drug and the body are conveniently divided into two classes. The actions of the drug on the body are termed pharmacodynamicprocesses.These properties determine the group in which the drug is classified, and they play the major role in deciding whether that group is appropriate therapy for a particular symptom or disease. -

Pharmacodynamics Drug Receptor Interactions Part-2
Pharmacodynamics: (Drug Receptor Interactions, Part 2) ………………………………………………………………………………………………………………………………………………………………………………………………………………… VPT: Unit I; Lecture-22 (Dated 03.12.2020) Dr. Nirbhay Kumar Asstt. Professor & Head Deptt. of Veterinary Pharmacology & Toxicology Bihar Veterinary College, Bihar Animal Sciences University, Patna Drug Receptor Interactions Agonist It is a drug that possesses affinity for a particular receptor and causes a change in the receptor that result in an observable effect. Full agonist: Produces a maximal response by occupying all or a fraction of receptors. (Affinity=1, Efficacy=1) Partial agonist: Produces less than a maximal response even when the drug occupies all of the receptors. (Affinity=1, Efficacy= 0 to 1) Inverse agonist: Activates a receptor to produce an effect in the opposite direction to that of the well recognized agonist. (Affinity=1, Efficacy= –1 to 0). Source: Rang & Dale’s Pharmacology, Elsevier Source: Good & Gilman’s The Pharmacological Basis of Therapeutics, 13th Edn. Antagonist An antagonist is a drug that blocks the response produced by an agonist. Antagonists interact with the receptor or other components of the effector mechanism, but antagonists are devoid of intrinsic activity (Affinity=1, Efficacy=0). Antagonist contd… Competitive Antagonism: It is completely reversible; an increase in the concentration of the agonist in the bio-phase will overcome the effect of the antagonist. Example: Atropine (Antimuscarinic agent) Diphenhydramine (H1 receptor blocker) Non-competitive antagonism: The agonist has no influence upon the degree of antagonism or its reversibility. Example: Platelet inhibiting action of aspirin (The thromboxane synthase enzyme of platelets is irreversibly inhibited by aspirin, a process that is reversed only by production of new platelets). -

Measuring Ligand Efficacy at the Mu- Opioid Receptor Using A
RESEARCH ARTICLE Measuring ligand efficacy at the mu- opioid receptor using a conformational biosensor Kathryn E Livingston1,2, Jacob P Mahoney1,2, Aashish Manglik3, Roger K Sunahara4, John R Traynor1,2* 1Department of Pharmacology, University of Michigan Medical School, Ann Arbor, United States; 2Edward F Domino Research Center, University of Michigan, Ann Arbor, United States; 3Department of Pharmaceutical Chemistry, School of Pharmacy, University of California San Francisco, San Francisco, United States; 4Department of Pharmacology, University of California San Diego School of Medicine, La Jolla, United States Abstract The intrinsic efficacy of orthosteric ligands acting at G-protein-coupled receptors (GPCRs) reflects their ability to stabilize active receptor states (R*) and is a major determinant of their physiological effects. Here, we present a direct way to quantify the efficacy of ligands by measuring the binding of a R*-specific biosensor to purified receptor employing interferometry. As an example, we use the mu-opioid receptor (m-OR), a prototypic class A GPCR, and its active state sensor, nanobody-39 (Nb39). We demonstrate that ligands vary in their ability to recruit Nb39 to m- OR and describe methadone, loperamide, and PZM21 as ligands that support unique R* conformation(s) of m-OR. We further show that positive allosteric modulators of m-OR promote formation of R* in addition to enhancing promotion by orthosteric agonists. Finally, we demonstrate that the technique can be utilized with heterotrimeric G protein. The method is cell- free, signal transduction-independent and is generally applicable to GPCRs. DOI: https://doi.org/10.7554/eLife.32499.001 *For correspondence: [email protected] Competing interests: The authors declare that no Introduction competing interests exist. -
![Response Vs. Log [L] - Full Agonist](https://docslib.b-cdn.net/cover/7019/response-vs-log-l-full-agonist-937019.webp)
Response Vs. Log [L] - Full Agonist
DavidsonX – D001x – Medicinal Chemistry Chapter 5 – Receptors Part 2 – Ligands Video Clip – Ligand Types Ligands can have different effects on a receptor. Each type of ligand can be readily classified according to its behavior. A type of ligand is the full agonist. The term agonist refers to a compound that binds a receptor and elicits a response (E). Full agonists elicit the same level of full response (E = Emax = 100%) as the endogenous ligand of the receptor. Graphically, a receptor-ligand interaction is plotted as response (E/Emax) vs. log [L]. The relationship is sigmoidal. A full agonist approaches full response (E/Emax = 1.0) as log [L] reaches relatively high levels. response vs. log [L] - full agonist 1 0.9 0.8 0.7 x a 0.6 m E 0.5 / E 0.4 0.3 0.2 0.1 0 log [L] Two ligands can achieve a full response without being equivalent. Ligands can differ with respect to the concentration required to trigger a response. A ligand that affects a response at a lower concentration has a higher potency. Potencies are measured as the effective ligand concentration required to reach a 50% response – EC50 or, in these graphs, log [EC50]. A more potent ligand has a lower EC50 value. full agonist comparison 1 0.9 full agonist 1 0.8 (more potent) full agonist 2 0.7 (less potent) x a 0.6 log EC m 50 E 0.5 / E 0.4 0.3 log EC 0.2 50 0.1 0 log [L] Partial agonists also cause a response, but they cannot reach the same, 100% response level of the endogenous ligand. -

Anew Drug Design Strategy in the Liht of Molecular Hybridization Concept
www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 “Drug Design strategy and chemical process maximization in the light of Molecular Hybridization Concept.” Subhasis Basu, Ph D Registration No: VB 1198 of 2018-2019. Department Of Chemistry, Visva-Bharati University A Draft Thesis is submitted for the partial fulfilment of PhD in Chemistry Thesis/Degree proceeding. DECLARATION I Certify that a. The Work contained in this thesis is original and has been done by me under the guidance of my supervisor. b. The work has not been submitted to any other Institute for any degree or diploma. c. I have followed the guidelines provided by the Institute in preparing the thesis. d. I have conformed to the norms and guidelines given in the Ethical Code of Conduct of the Institute. e. Whenever I have used materials (data, theoretical analysis, figures and text) from other sources, I have given due credit to them by citing them in the text of the thesis and giving their details in the references. Further, I have taken permission from the copyright owners of the sources, whenever necessary. IJCRT2012039 International Journal of Creative Research Thoughts (IJCRT) www.ijcrt.org 284 www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 f. Whenever I have quoted written materials from other sources I have put them under quotation marks and given due credit to the sources by citing them and giving required details in the references. (Subhasis Basu) ACKNOWLEDGEMENT This preface is to extend an appreciation to all those individuals who with their generous co- operation guided us in every aspect to make this design and drawing successful. -

Volume C, Module 2 Opioids: Basics of Addiction; Treatment with Agonists, Partial Agonists, and Antagonists
VolumeVolume C,C, ModuleModule 22 Opioids:Opioids: BasicsBasics ofof Addiction;Addiction; TreatmentTreatment withwith Agonists,Agonists, PartialPartial Agonists,Agonists, andand AntagonistsAntagonists Treatnet Training Volume C: Module 2 – Updated 18 October 2007 ModuleModule 2:2: TrainingTraining goalsgoals ToTo describedescribe the:the: ¾ KeyKey componentscomponents ofof opiateopiate addictionaddiction andand itsits medicalmedical // psychiatricpsychiatric consequencesconsequences ¾ BenefitsBenefits andand limitationslimitations ofof methadonemethadone asas aa pharmacotherapypharmacotherapy forfor opiateopiate dependencedependence ¾ BenefitsBenefits andand limitationslimitations ofof buprenorphinebuprenorphine asas aa pharmacotherapypharmacotherapy forfor opiateopiate dependencedependence ¾ BenefitsBenefits andand limitationslimitations ofof narcoticnarcotic antagonistsantagonists forfor overdoseoverdose (naloxone)(naloxone) andand relapserelapse preventionprevention (naltrexone)(naltrexone) forfor opiateopiate dependencedependence ModuleModule 2:2: WorkshopsWorkshops WorkshopWorkshop 1:1: Opiates:Opiates: WhatWhat theythey are,are, problemsproblems associatedassociated withwith theirtheir use,use, andand medicalmedical treatmenttreatment implicationsimplications WorkshopWorkshop 2:2: OpiateOpiate addictionaddiction treatmenttreatment withwith methadonemethadone WorkshopWorkshop 3:3: OpiateOpiate addictionaddiction treatmenttreatment withwith buprenorphinebuprenorphine WorkshopWorkshop 4:4: OpiateOpiate AntagonistAntagonist Treatment:Treatment: -

International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology
0031-6997/03/5504-597–606$7.00 PHARMACOLOGICAL REVIEWS Vol. 55, No. 4 Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 30404/1114803 Pharmacol Rev 55:597–606, 2003 Printed in U.S.A International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology RICHARD R. NEUBIG, MICHAEL SPEDDING, TERRY KENAKIN, AND ARTHUR CHRISTOPOULOS Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (R.R.N.); Institute de Recherches Internationales Servier, Neuilly sur Seine, France (M.S.); Systems Research, GlaxoSmithKline Research and Development, Research Triangle Park, North Carolina (T.K.); and Department of Pharmacology, University of Melbourne, Parkville, Australia (A.C.) Abstract ............................................................................... 597 I. Introduction............................................................................ 597 II. Working definition of a receptor .......................................................... 598 III. Use of drugs in definition of receptors or of signaling pathways ............................. 598 A. The expression of amount of drug: concentration and dose ............................... 598 Downloaded from 1. Concentration..................................................................... 598 2. Dose. ............................................................................ 598 B. General terms used to describe drug action ........................................... -
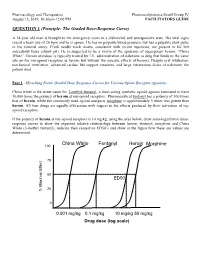
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined. -

1.11 Antidepressants and the Gut Microbiota
UCC Library and UCC researchers have made this item openly available. Please let us know how this has helped you. Thanks! Title Effects of psychotropic drugs on the microbiota-gut-liver-brain axis Author(s) Cussotto, Sofia Publication date 2019 Original citation Cussotto, S. 2019. Effects of psychotropic drugs on the microbiota-gut- liver-brain axis. PhD Thesis, University College Cork. Type of publication Doctoral thesis Rights © 2019, Sofia Cussotto. http://creativecommons.org/licenses/by-nc-nd/3.0/ Item downloaded http://hdl.handle.net/10468/9468 from Downloaded on 2021-09-23T15:30:09Z Ollscoil na hÉireann, Corcaigh National University of Ireland, Cork Department of Anatomy and Neuroscience Head of Dept. John F. Cryan Effects of Psychotropic Drugs on the Microbiota-Gut-Liver-Brain Axis Thesis presented by Sofia Cussotto, MPharm Under the supervision of Prof. John F. Cryan Prof. Timothy G. Dinan For the degree of Doctor of Philosophy June 2019 Table of Contents Declaration ......................................................................................................... IV Author Contributions .......................................................................................... IV Acknowledgments ............................................................................................... V Publications and presentations ........................................................................... VI Abstract ............................................................................................................ VIII -

PHARMACOGENOMICS Know Your GPCR Mutations (And Target Them Right)
RESEARCH HIGHLIGHTS Nature Reviews Drug Discovery | Published online 1 Feb 2018; doi:10.1038/nrd.2018.13 mangsaab/iStock/Getty Images Plus PHARMACOGENOMICS Know your GPCR mutations (and target them right) Natural human genetic variations may within known functional sites, such MOR. Given the huge number of cause patients to respond differently as ligand binding, effector binding or drugs targeting the MOR that are to the same medication. Therefore, post-translational modification sites. prescribed, if even a small fraction understanding genetic variation in To explore the functional are ineffective or cause adverse drug targets can maximize efficacy implications of such mutations, the reactions, the economic burden and reduce side effects. In a new study, authors then experimentally analysed may be considerable. a team led by M. Madan Babu the impact of three MVs near the However, “as it stands, no receptor present a comprehensive analysis of ligand-binding pocket of the μ-opioid variants are included in the labelling G protein-coupled receptor (GPCR) receptor (MOR) on G protein activa- information of any GPCR drug genetic variants and find that GPCRs tion by the endogenous agonist endo- target by any regulatory agency,” says targeted by marketed drugs show morphine 1, morphine (full agonist), Alexander Hauser, first author of this genetic variation within functional buprenorphine (partial agonist) and study. “This is likely going to change regions such as drug-binding sites. naloxone (antagonist). Whereas one with increased sequencing efforts, GPCRs mediate the therapeutic variant (M153V) resulted in partial bigger cohorts and better character- effects of more than 30% of FDA- LoF and reduced the response to both ization of the variants”. -

Introduction to Pharmacodynamics Reza Karimi 6
CHAPTER Introduction to Pharmacodynamics Reza Karimi 6 1. Understand the physiology behind the gastrointestinal tract and the route of oral drug administration and VES physiological influences on pharmacodynamics. I 2. Understand the dynamics and functions of the major signal transduction systems and their different biomedi- cal and biological responses in regard to receptor–ligand interactions. 3. Learn about the dynamics and mathematical expressions behind receptor–ligand interactions. OBJECT 4. Understand dose–response relationships and factors that affect a pharmacological response. 5. Learn about agonistic, antagonistic, and partial agonistic binding of drugs to receptors. 6. Learn about different concepts such as addition, synergism, and potentiation that lead to an enhancement effect of drugs. 7. List a few regulatory mechanisms for receptors. 8. Implement a series of Learning Bridge assignments at your experiential sites to bridge your didactic learning with your experiential experiences. 1. cAMP: cyclic adenosine 3' ,5''-monophosphate; a second messenger that plays an important role in signal NS transduction. IO T 2. cGMP: cyclic guanosine 3' ,5''monophosphate; a second messenger that plays an important role in signal I N transduction. I 3. Dose–response relationship: when an endogenous or exogenous ligand binds to a receptor and produces a EF D pharmacological effect. The effect can approach a maximum value (also called Emax) in which a further increase in the ligand concentration does not produce any higher response. 4. Efficacy: the ability of a drug to produce a pharmacological response when it interacts with its receptor. 5. First-pass metabolism: a type of metabolism in which drugs that are absorbed by the gastrointestinal tract go through the portal vein to the liver and are metabolized there before they are distributed to the general ERMS AND AND ERMS circulation. -

For the Degree of DOCTOR of PHILOSOPHY Science Faculty
SYNTHESIS OF BIOLOGICALLY ACTIVE NATURAL PRODUCTS AND PHARMACOLOGICALLY ACTIVE MOLECULES BY COMBINATORIAL CHEMISTRY A THESIS Submitted to the SHIVAJI UNIVERSITY, KOLHAPUR For the Degree of DOCTOR OF PHILOSOPHY in CHEMISTRY Science Faculty By ANIL M. DESHPANDE Under the Guidance of Dr. A. A. NATU Division of Organic Chemistry (Synthesis) National Chemical Laboratory Pune 411 008 SEPTEMBER 2001 DEDICATED To My Parents For Their Warmth, Humor And Ethics CERTIFICATE This is to certify that the thesis entitled “SYNTHESIS OF BIOLOGICALLY ACTIVE NATURAL PRODUCTS AND PHARMACOLOGICALLY ACTIVE MOLECULES BY COMBINATORIAL CHEMISTRY” which is being submitted herewith for the award of the Degree of Philosophy in Chemistry of Shivaji University, Kolhapur is the result of the original research work completed by Mr. Anil M. Deshpande under my supervision and guidance at the National Chemical Laboratory, Pune and to the best of my knowledge and belief the work embodied in this thesis has not formed earlier the basis for the award of any Degree or similar title of this or any other University or examining body. (A. A. Natu) Research Guide September 2001 Scientist, Division of Organic Chemistry (Synthesis) National Chemical Laboratory Pune 411 008. DECLARATION I hereby declare that the thesis entitled “SYNTHESIS OF BIOLOGICALLY ACTIVE NATURAL PRODUCTS AND PHARMACOLOGICALLY ACTIVE MOLECULES BY COMBINATORIAL CHEMISTRY” completed and written by me has not previously formed the basis for the award of any Degree or Diploma or other similar title of this or any other University or examining body. (Anil M. Deshpande) Division of Organic Chemistry (Synthesis) September 2001 National Chemical Laboratory Pune 411 008.