1 Additional File 1. Supplemental Figures S1-S6: a Trans Locus Causes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Untergrundbahnbau Frankfurt Am Main FH Potsdam Philipp Holzmann Archiv Prof
Vitali Elin Untergrundbahnbau Frankfurt am Main FH Potsdam Philipp Holzmann Archiv Prof. Dr. phil. A. Kahlow Untergrundbahnbau Frankfurt am Main vom Fachbereich Bauingenieurwesen der Fachhochschule Potsdam zur Erlangung des Leistungsnachweises im Ingenieurprojekt: „Bilderarchiv der Philipp Holzmann AG“ Vitali Elin Gutachter: Prof. Dr. phil. A. Kahlow Potsdam, Januar 2017 1 Vitali Elin Untergrundbahnbau Frankfurt am Main FH Potsdam Philipp Holzmann Archiv Prof. Dr. phil. A. Kahlow Inhaltsverzeichnis 1. Allgemeines ….......................................................................................................... 3 2. Geschichte der Frankfurter U-Bahn …..................................................................... 6 3. Bauweisen ….......................................................................................................... 12 3.1 Tunnelbauten …................................................................................................. 12 4. Streckennetz …....................................................................................................... 15 4.1. Strecke A …..................................................................................................... 16 4.2. Strecke C …..................................................................................................... 19 5. Kosten …................................................................................................................. 20 6. Quellenverzeichnis …............................................................................................ -

Using Shapley Additive Explanations to Interpret Extreme Gradient Boosting Predictions of Grassland Degradation in Xilingol, China
Geosci. Model Dev., 14, 1493–1510, 2021 https://doi.org/10.5194/gmd-14-1493-2021 © Author(s) 2021. This work is distributed under the Creative Commons Attribution 4.0 License. Using Shapley additive explanations to interpret extreme gradient boosting predictions of grassland degradation in Xilingol, China Batunacun1,2, Ralf Wieland2, Tobia Lakes1,3, and Claas Nendel2,3 1Department of Geography, Humboldt-Universität zu Berlin, Unter den Linden 6, 10099 Berlin, Germany 2Leibniz Centre for Agricultural Landscape Research (ZALF), Eberswalder Straße 84, 15374 Müncheberg, Germany 3Integrative Research Institute on Transformations of Human-Environment Systems, Humboldt-Universität zu Berlin, Friedrichstraße 191, 10099 Berlin, Germany Correspondence: Batunacun ([email protected]) Received: 25 February 2020 – Discussion started: 9 June 2020 Revised: 27 October 2020 – Accepted: 10 November 2020 – Published: 16 March 2021 Abstract. Machine learning (ML) and data-driven ap- Land-use change includes various land-use processes, such proaches are increasingly used in many research areas. Ex- as urbanisation, land degradation, water body shrinkage, and treme gradient boosting (XGBoost) is a tree boosting method surface mining, and has significant effects on ecosystem ser- that has evolved into a state-of-the-art approach for many vices and functions (Sohl and Benjamin, 2012). Grassland is ML challenges. However, it has rarely been used in sim- the major land-use type on the Mongolian Plateau; its degra- ulations of land use change so far. Xilingol, a typical re- dation was first witnessed in the 1960s. About 15 % of the gion for research on serious grassland degradation and its total grassland area was characterised as being degraded in drivers, was selected as a case study to test whether XG- the 1970s, which rose to 50 % in the mid-1980s (Kwon et Boost can provide alternative insights that conventional land- al., 2016). -

Stadtbahn Hannover
Stadtbahn mit Haltestelle DB City-Ticket GVH Kombiticket 3 Bremen Soltau Tram line with stop gültig in Zone gültig in den Zonen Stadtbahn Hannover Stadtbahn mit Tunnelstation RE 1 valid in zone A valid in zones A B C RB38 Underground station RE 8 Bennemühlen Uelzen Veranstaltungslinie Hamburg 16 18 Special service S4 RE2 Abweichender Fahrweg Nienburg Hannover Flughafen / / / RE3 10 RE2 RE3 n n n im Nachtsternverkehr S2 S5 Celle Night service S6 RegionalExpress 5 Stöcken 5 Langenhage Langenhage Langenhage 1 Langenhagen 3 Altwarmbüchen RE 2 Marshof S S7 t/ Langenforther Zentrum Platz Kurt-Schumacher-Allee RB 38 RegionalBahn 4 S Langenhagen/Angerstr. Regional train 2 S S-Bahn Stöckener Markt Berliner Platz S2 8 4 Auf der HorsAuf der Horst/SkorpiongasseSchönebeckerPascalstr. AlleeWissenschaftsparkJädekampAuf Marienwerde der KlappenburgLauckerthofr Altwarmbüchen/ Garbsen 6 Nordhafen 2 Alte Heide Suburban train Zentrum Übergang zum Bus Wiesenau 1 RE Mecklenheidestr. 9 Interchange with bus Freudenthalstr. Bahnstrift Fasanenkrug Altwarmbüchen/ 38 Alter Flughafen Ernst-Grote-Str. Park+Ride Beneckeallee B RE Tempelhofweg Stufenfreier Zu-/Abgang Stadtfriedhof Stöcken Friedenauer Str. Zehlendorfweg Stadtfriedhof Step-free access Fuhsestr. Kabelkamp Bothfeld Altwarmbüchen/ RE1 RE8 RE60 Fuhse- Krepenstr. Papenwinkel Opelstr. Tarifzonen RE70 S1 S2 RE1 RE8 RE60 RE70 S1 S2 S51 str./Bhf. 2 RE3 R Kurze-Kamp-Str. A B C E Windausstr. Vahrenheider Markt Fare zones R Wunstorf Seelze Bahnhof Leinhausen Hainhölzer Reiterstadion Bothfeld Oldenburger Allee Markt Büttnerstr. S51 Bothfelder Kirchweg 7 Stand: Oktober 2020 Herrenhäuser Markt Großer Kolonnenweg gvh.de S Fenskestr. Niedersachsenring Buchholz/Bhf. Stadtfriedhof Lahe 6 S Schaumburgstr. Dragonerstr. 8 Haltenhoffstr. Bahnhof C B A Herrenhäuser Gärten Nordstadt Vahrenwalder Platz 11 Appelstr. -

Rail Transport in the World's Major Cities
Feature Evolution of Urban Railways (part 2) Rail Transport in The World’s Major Cities Takao Okamoto and Norihisa Tadakoshi Many of the world’s large cities grew in development, particularly with regard to American city; and Hong Kong, Seoul and conjunction with railways, and today, the following: Tokyo representing Asian cities. large cities cannot depend only on motor • The correlation between railways and vehicles for transportation. With worries urban growth over global environmental issues, public • The location of terminals for intercity London transportation systems are increasingly and intra-city transport seen as an important way to expand and • Examination of various public transit Located in southeast England near the revitalize large cities, while consuming systems, including non-infrastructure mouth of the River Thames, London less energy and other resources. • Strategic planning of rail networks expanded during the Industrial Revolution This article looks at public transportation based on urban development trends (1760–1850) and secured its dominance systems in some major cities of the world and future models as the heart of the British Empire during and identifies similarities and differences The cities selected for this comparative the Victorian era (1837–1901). It’s in areas such as history of development, study are: London as the first city to adapt population grew from about 500,000 in railway networks, and method of rail technology to public transport; Paris, the 17th century to 4.5 million by the end operation. Our aim is to explore the future Berlin and Moscow as three major of the 19th century. The modern relationship between urban and transport European cities; New York as a North- metropolis of Greater London consists of The London Railway Network King’s Cross/St.Pancras Paddington LCY Victoria Waterloo LHR R. -

Und Informations-Dialog 2018 Baumaßnahmen Im Netz Der Berliner S-Bahn 2018 - 2020
2. Bau- und Informations-Dialog 2018 Baumaßnahmen im Netz der Berliner S-Bahn 2018 - 2020 DB Netz AG | I.NP-O-D-BLN(BS) + I.NM-O-F(S) | Berlin | 17.07.2018 Übersicht Regionalbereich Ost – Netz Berliner S-Bahn Bauschwerpunkte 2018 Ersatzneubau SÜ Rhinstraße + ESTW+ZBS S7 Ost SEV Lichtenberg–Springpfuhl/ Wuhletal Wochenenden April bis Juni + Dezember 2018 Brückenarbeiten S2 Nord + Neubau SÜ BAB114 SEV Lichtenberg–Ahrensfelde/ Wartenberg SEV Blankenburg–Karow + Blankenburg- 19.10.–25.10.2018 Schönfließ SEV Sprinpfuhl–Wartenberg/ Ahrensfelde 26.06.–16.07.2018 20.07.–23.07.2018 SEV Blankenburg–Buch + Blankenburg- Schönfließ 16.07.–23.07.2018 SEV Blankenburg–Buch Ersatzneubau 23.07.–17.08.2018 EÜ Thälmannstr. + Entflechtung S-/F- ZBS S5 West Schienenauswechslung Bahn Bf Strausberg SEV Westkreuz–Spandau SEV Tiergarten–Charlottenburg SEV Mahlsdorf– 13.08.–16.08.2018 (in Prüfung 23.07.-03.08.2017 Strausberg Nord Verschiebung IBN nach 01/2019) 23.11.–29.11.2018 Umbau Ostkreuz – Neubau Bahnsteig Karlshorst Ibn Endzustand 4-gleisigkeit SEV Rummelsburg–Wuhlheide ZBS S7 West + Begegnungsabschn. Potsdam SEV Ostkreuz–Karlshorst 06.07.–16.07.2018 SEV Wannsee–Potsdam 02.11.–12.11.2018 kein Verkehrshalt Karlshorst 03.08.–06.08. + 10.08.–13.08. + 14.12.–17.12.2018 SEV Alexanderplatz–Lichtenberg 06.07. – 06.08.2018 SEV Westkreuz–Wannsee + Babelsberg–Potsdam 02.11.–05.11. + 09.11.–12.11.2018 eingleisig Wuhlheide – Karlshorst 31.08.–03.09.2018 06.08. – 15.08.2018 SEV Westkreuz–Grunewald 16.11.–19.11.2018 Ende Neubau EÜ Sterndamm + Neubau PT Schöneweide bis 2021 bis 20.08.2018 halbseitg. -

X-Ray Structural Analyses of Cyclodecasulfur (S10) and of A
X-Ray Structural Analyses of Cyclodecasulfur (S10) and of a Cyclohexasulfur-Cyclodecasulfur Molecular Addition Compound (S6 • S10) [1] Ralf Steudel*, Jürgen Steidel Institut für Anorganische und Analytische Chemie, Technische Universität Berlin, Sekr. C2, D-1000 Berlin 12 Richard Reinhardt** Institut für Kristallographie der Freien Universität Berlin, Takustraße 6, D-1000 Berlin 33 Dedicated to Professor Dr. Karl Winnacker on the occasion of his 80th birthday Z. Naturforsch. 38 b, 1548-1556 (1983); received August 11, 1983 Elemental Sulfur, Sulfur Rings, Molecular Structure, Crystal Structure, Vibrational Spectra Low temperature X-ray structural analyses of monoclinic single crystals of Sio and Sö • Sio (prepared from the components) show that the cyclic Sio molecule exhibits the same D2 conformation in both compounds with bond distances between 203.3 and 208.0pm, bond angles (a) between 103 and 111°, and torsional angles (r) between 73 and 124°. The Sß molecule (site symmetry Ci) in Sö • Sio is very similar to the one in pure Sö (dss = 206.2 pm, a= 103°, r = 74°). All intermolecular interactions are of van-der-Waals type. The Raman spectrum of S6 • Sio can be explained by a superposition of the Se and Sio spectra. Introduction In all cases the reason for the differing bond distances is the variation of the torsional angles The tremendous industrial importance of ele- leading to a varying degree of lone-pair-lone-pair mental sulfur and its surplus expected for the near repulsion between neighboring atoms. In fact, the future (and occasionally already experienced in the torsional angles r in sulfur rings Sre vary between 0° past) have stimulated considerable research ac- and 140° resulting in a bond length variation of tivities with the result that 19 well characterized 19 pm or 9% [18]. -

Techno-Economic Assessment of Battery Electric Trains and Recharging Infrastructure Alternatives Integrating Adjacent Renewable Energy Sources
sustainability Article Techno-Economic Assessment of Battery Electric Trains and Recharging Infrastructure Alternatives Integrating Adjacent Renewable Energy Sources Christoph Streuling 1,* , Johannes Pagenkopf 1, Moritz Schenker 1 and Kim Lakeit 2 1 German Aerospace Center (DLR), Institute of Vehicle Concepts, 70569 Stuttgart, Germany; [email protected] (J.P.); [email protected] (M.S.) 2 Institute of Electric Power Systems, Otto von Guericke University, 39106 Magdeburg, Germany; [email protected] * Correspondence: [email protected] Abstract: Battery electric multiple units (BEMU) are an effective path towards a decarbonized regional rail transport on partly electrified rail lines. As a means of sector coupling, the BEMU recharging energy demand provided through overhead line islands can be covered from decentralized renewable energy sources (RES). Thus, fully carbon-free electricity for rail transport purposes can be obtained. In this study, we analyze cost reduction potentials of efficient recharging infrastructure positioning and the feasibility of covering BEMU energy demand by direct-use of locally produced renewable electricity. Therefore, we set up a model-based approach which assesses relevant lifecycle costs (LCC) of different trackside electrification alternatives comparing energy supply from local RES and grid consumption. The model-based approach is applied to the example of a German regional rail line. In Citation: Streuling, C.; Pagenkopf, J.; the case of an overhead line island, the direct-use of electricity from adjacent wind power plants with Schenker, M.; Lakeit, K. on-site battery storage results in relevant LCC of EUR 173.4 M/30a, while grid consumption results in Techno-Economic Assessment of EUR 176.2 M/30a whereas full electrification results in EUR 224.5 M/30a. -

World Para Swimming World Records Long Course
World Para Swimming World Records Long Course World Para Swimming World Records created by IPC Sport Data Management System Gender: Men | Course: Long Course Men's 50 m Freestyle Class Name NPC Birth Time Date City Country S1 Mamistvalov, Itzhak ISR 1979 01:03.80 2014-03-09 Esbjerg Denmark S2 Zou, Liankang CHN 1995 00:50.65 2016-09-11 Rio de Janeiro Brazil S3 Huang, Wenpan CHN 1995 00:38.81 2017-12-06 Mexico City Mexico S4 Leslie, Cameron NZL 1990 00:37.14 2019-09-13 London Great Britain S5 Fantin, Antonio ITA 2001 00:30.16 2019-06-01 Lignano Sabbiadoro Italy S6 Xu, Qing CHN 1992 00:28.57 2012-09-04 London Great Britain S7 Trusov, Andrii UKR 2000 00:27.07 2019-09-15 London Great Britain S8 Tarasov, Denis RUS 1993 00:25.32 2014-08-10 Eindhoven Netherlands S9 Barlaam, Simone ITA 2000 00:24.00 2019-09-15 London Great Britain S10 Brasil, Andre BRA 1984 00:23.16 2012-08-31 London Great Britain S11 Yang, Bozun CHN 1986 00:25.27 2012-09-01 London Great Britain S12 Veraksa, Maksym UKR 1984 00:22.99 2009-10-21 Reykjavik Iceland S13 Boki, Ihar BLR 1994 00:23.20 2015-07-13 Glasgow Great Britain IPC Sport Data Management System Page 1 of 22 29 September 2021 at 00:11:44 CEST World Para Swimming World Records Long Course Men's 100 m Freestyle Class Name NPC Birth Time Date City Country S1 Mamistvalov, Itzhak ISR 1979 02:15.83 2012-09-01 London Great Britain S2 Liu, Benying CHN 1996 01:46.63 2016-09-11 Rio de Janeiro Brazil S3 Lopez Diaz, Diego MEX 1994 01:32.69 2018-06-08 Berlin Germany S4 Dadaon, Ami Omer ISR 2000 01:19.77 2021-05-17 Funchal Portugal -

Stadt Zürich | Zurich City
Stadt Zürich | Zurich City Zürich Flughafen Tarifzone 110 111 121 Fare zone 110 S9 REGENSDORF 10 S Glattbrugg 762 116 111 1 S24 768 Halden 5 764 12 KATZENSEE 110 Bhf. Lätten- 115 162 Käshaldenstr. Im Ebnet S16 Zentrumwiesen 161 Birch-| 114 113 124 Köschenrüti Glatttalstr. S2 Post 160 118 163 Leimgrübelstr. 123 Schönauring 75 Ausser- 112 120 164 dorfstr. 117 S6 Mühlacker 761 S7 UnteraffolternSchwandenholzWaidhof 742 Ettenfeld 761 Frohbühl- 111 170 62 121 122 171 str. Bhf. Opfikon Chrüzächer Holzerhurd 184 Friedhof Seebach Giebeleichstr. 154 110 130 135 32 Schwandenholz Hertenstein- 131 172 Buhnstr. Lindbergh- OPFIKON 173 Aspholz 40 str. 140 Fronwald platz 155 132 Oberhusen 142 133 Blumen-feldstr. 150 Staudenbühl 141 134 111 Bahnhof 75 143 491 61 Affoltern Nord 768 Lindbergh-Allee 156 151 Hungerbergstr. Maillartstr.Stierenried 180 40 Himmeri Chavez-Allee 152 154 62 Glattpark 182 183 Bhf. Seebach Seebacherplatz Wright-Strasse 153 181 Zehntenhausplatz S6 S21 Fernsehstudio Glaubtenstr.Glaubtenstr. Einfangstr. Hürstholz 12 ENGSTRINGEN 64 Neun- 111 110 Mötteliweg Felsenrain- Grünwald brunnen str. Genossen- 11 Bollinger- schaftsstr. Schauen- berg weg 14 Oerlikerhus Orionstr. Heizenholz Bhf. Auzelg Oerlikon Leutschen- Lerchenhalde Glaubtenstr. Riedbach Rütihof Lerchen- weg Nord bach WALLISELLEN Süd 62 Chalet- 781 rain 79 Herti Belair Giblenstr. Hagenholz Auzelg Ost 46 Schumacherweg Bahnhof 485 80 Geeringstr. Schützenhaus Bhf. Höngg Birchstr. Wallisellen 80 Oerlikon S8 S14 S19 Riedhofstr. Neu- Platz 61 Ost 37 Oberwiesen-Max-Bill- Saatlenstr. S8 Friedhof Bahnhof Hallenbad Riedgraben Dreispitz Aubrücke 89 affoltern str. Hönggerberg Oerlikon 94 Oerlikon 94 Flora- 40 str. 308 Ifang 62 Schürgi- 304 Michelstr. Althoos Frankental ETH 61 str. 110 121 Segantinistr.Singlistr. -

Train Simulator - Right Through Berlin
Train Simulator - Right through Berlin © Historical S-Bahn Berlin e.V. 2020 Page 1 Train Simulator - Right through Berlin Manual "Mitten durch Berlin" - Operation Series 476 Table of contents Table of contents ....................................................................................................... 2 0. authors ................................................................................................................ 3 1. introduction .......................................................................................................... 3 2. system requirements .............................................................................................. 4 3. the driver's cab ..................................................................................................... 5 4. upgrading .......................................................................................................... 10 4.1 Train is cold (full upgrade) .............................................................................. 10 4.2 Transfer of trains from other Tf ......................................................................... 10 4.3 Train is fully upgraded .................................................................................... 10 4.4 Change of driver's cab ................................................................................... 11 5. door control ....................................................................................................... 11 6. driving and braking ........................................................................................... -
Buses and Trains In
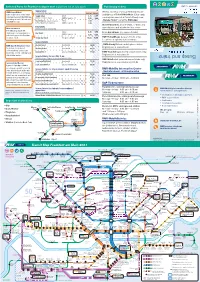
Selected Fares for Frankfurt & airport 2021 (valid from 1st of July 2021) Purchasing tickets 1.7.2021 from valid and Tickets and and Tickets and s Transit Maps Maps Transit Transit Single ticket Weekly, monthly or annual RMV tickets are Maps Transit RMV-PrepaidRabatt For immediate travel only Frankfurt & Airport Save 20 percent on every single available as eTicket RheinMain (Chip cards Single ticket Adults 2,75 5,10 ticket purchase with the RMV-App Valid for 1 trip incl. change Children aged 6 - 14 1,55 3,00 can be purchased at all ticket offices) or as by loading at least € 40 onto your Adults „Handy-Ticket“ using the RMV-App meinRMV account. Short-trip Max. 2km, destinations 1,50 listed at stops/ticket machines Children aged 6 - 14 1,00 Ticket machines at all S-Bahn, U-Bahn and Day tickets tram stations and at selected bus stops Ideal for families! Valid until 5 am on following day The RMV group day ticket Adults 5,35 9,95 entitles up to five passengers to Day ticket From bus drivers (no season tickets) Children aged 6 - 14 unlimited travel within Frankfurt 3,00 5,85 for only € 11,50. Group day ticket For up to 5 people 11,50 16,95 VGF-TicketShops (season tickets only) Locations at vgf-ffm.de/ticketshops Season tickets „RMV-HandyTicket“ mobile phone ticket RMV-App: Mobile phone ticket Weekly ticket transferable 26,80 Registration at www.rmv.de 2021 2021 2021 No change, no queues at Monthly ticket transferable 93,10 2021 in one in in one in ticket-machines, and no fare- Annual ticket personal or transferable 12 × 77,60 RMV-TicketShop (selected season tickets only) knowledge needed. -

Berlin 2018 World Para Swimming World Series PI Classification Schedule Version 1.2
Berlin 2018 World Para Swimming World Series PI Classification Schedule Version 1.2 Athletes: Must present to the Classification 15 minutes before the allocated time on the classification schedule. Must bring a passport or another official photo form of identification to classification. Will be required to read and sign a classification consent form prior to presenting to the classification panel. Must be accompanied by one support staff from their team. Must bring their own interpreter if they do not speak English. 4-Jun-18 Time SDMS ID NPC Family Name Given Name S SB SM Status 9:00 29994 TUR Inci Omer Onur S4 New 9:00 19482 ISR Gordiychuk Yuliya S9 SB8 SM9 Review 9:00 4201 BLR Shavel Natalia S5 SB4 SM5 Review 9:00 38962 ESP Cortes Melgar Jaume S5 SB4 SM5 New 10:30 33328 TUR Dogan Ismail Can S5 SB4 SM5 Review 10:30 23573 KAZ Yeldekenov Yerlan S9 SB8 New 10:30 22473 ISR Malyar Ariel S5 SB4 SM5 Review 10:30 35895 CZE Fryba Petr S8 New 12:15 15071 ESP Rolo Marichal Judit S7 SB7 SM7 Review 12:15 5338 AUT Onea Andreas S8 SB8 SM8 Review 12:15 26226 BLR Shchalkanau Yahor S10 SB9 SM10 Review Lunch Break 13:15 14:15 35154 KAZ Pronkina Viktoriya S7 New 14:15 19472 ESP Rodriguez Martinez Ines Rocio S4 SB3 SM3 New 14:15 22955 CZE Andrysek Petr S6 SB5 SM6 Review 14:15 26225 BLR Musarau Artsiom S7 SB6 SM7 Review 16:00 11498 EST Tilk Brenda S7 N/A N/A Review 16:00 20050 EST Topkin Matz S5 SB4 SM5 Review 16:00 17934 CZE Duskova Vendula S8 SB7 SM8 Review 16:00 22511 BLR Svadkouskaya Aliaksandra S10 SB9 SM10 Review 17:30 5942 ESP Gascon Sarai S9 SB9 SM9