DYNC1H1 Mutations Associated with Neurological Diseases Compromise
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

HOOK3 Is a Scaffold for the Opposite-Polarity Microtubule-Based
bioRxiv preprint doi: https://doi.org/10.1101/508887; this version posted December 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. HOOK3 is a scaffold for the opposite-polarity microtubule-based motors cytoplasmic dynein and KIF1C Agnieszka A. Kendrick1, William B. Redwine1,2†, Phuoc Tien Tran1‡, Laura Pontano Vaites2, Monika Dzieciatkowska4, J. Wade Harper2, and Samara L. Reck-Peterson1,3,5 1Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, CA, 92093. 2 Department of Cell Biology, Harvard Medical School, Boston, MA 02115. 3Section of Cell and Developmental Biology, Division of Biological Sciences, University of California San Diego, La Jolla, CA 92093. 4Department of Biochemistry and Molecular Genetics, University of Colorado Denver, Aurora, CO 80045. 5Howard Hughes Medical Institute †Present address: Stowers Institute for Medical Research, Kansas City, MO 64110 ‡Present address: Department of Molecular and Cellular Biology, Harvard University, Cambridge, MA 02138. *Correspondence to: Samara Reck-Peterson 9500 Gilman Drive, Leichtag 482 La Jolla CA, 92093 [email protected]; https://orcid.org/0000-0002-1553-465X 1 bioRxiv preprint doi: https://doi.org/10.1101/508887; this version posted December 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Product Datasheet KIF1C Antibody NB100-57510
Product Datasheet KIF1C Antibody NB100-57510 Unit Size: 0.1 ml Store at 4C. Do not freeze. Reviews: 1 Publications: 1 Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NB100-57510 Updated 3/16/2021 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NB100-57510 Page 1 of 3 v.20.1 Updated 3/16/2021 NB100-57510 KIF1C Antibody Product Information Unit Size 0.1 ml Concentration 0.2 mg/ml Storage Store at 4C. Do not freeze. Clonality Polyclonal Preservative 0.09% Sodium Azide Isotype IgG Purity Immunogen affinity purified Buffer TBS and 0.1% BSA Product Description Host Rabbit Gene ID 10749 Gene Symbol KIF1C Species Human, Mouse Immunogen The immunogen recognized by this antibody maps to a region between residue 1053 and the C-terminus (residue 1103) of human kinesin family member 1C (lethal toxin sensitivity 1) using the numbering given in entry NP_006603.2 (GeneID 10749). Product Application Details Applications Western Blot, Immunoprecipitation Recommended Dilutions Western Blot 1:2000-1:10000, Immunoprecipitation 2 - 5 ug/mg lysate Page 2 of 3 v.20.1 Updated 3/16/2021 Images Western Blot: KIF1C Antibody [NB100-57510] - KIF1C is a novel HOOK3 -interacting protein.sfGFP-3xFLAG and full length (FL) HOOK3, HOOK3 (1-552), and HOOK3 (553-718) all tagged with sfGFP and 3xFLAG were immunoprecipitated with alpha-FLAG antibodies from transiently transfected HEK-293T cells. Western blots with alpha-HC, alpha- FAM160A2, alpha-KIF1C, and alpha-FLAG antibodies were used to determine which proteins co-immunoprecipitated with each HOOK3 construct.DOI:http://dx.doi.org/10.7554/eLife.28257.015 Image collected and cropped by CiteAb from the following publication (https://elifesciences.org/articles/28257), licensed under a CC-BY licence. -

047605V1.Full.Pdf
bioRxiv preprint doi: https://doi.org/10.1101/047605; this version posted April 8, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Assembly and Activation of Dynein-Dynactin by the Cargo Adaptor Protein Hook3 Courtney M. Schroeder1,2 and Ronald D. Vale1,2 1The Howard Hughes Medical Institute, University of California, San Francisco, San Francisco, California, USA 2Department of Cellular and Molecular Pharmacology, University of California, San Francisco, San Francisco, California, USA. Corresponding Author: Ronald D. Vale Dept. of Cellular and Molecular Pharmacology University of California, San Francisco Genentech Hall, MC 2200, Room N312A 600-16th Street San Francisco, CA 94158-2517 E-mail: [email protected] Phone: 415-476-6380 Fax: 415-514-4412 bioRxiv preprint doi: https://doi.org/10.1101/047605; this version posted April 8, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 2 Abstract Metazoan cytoplasmic dynein moves processively along microtubules with the aid of dynactin and an adaptor protein that joins dynein and dynactin into a stable ternary complex. Here, we have examined how Hook3, a cargo adaptor involved in Golgi and endosome transport, forms a motile dynein-dynactin complex. We show that the conserved Hook domain interacts directly with the dynein light intermediate chain 1 (LIC1). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer
Supplementary Materials: Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer Marcos Díaz-Gay, Sebastià Franch-Expósito, Coral Arnau-Collell, Solip Park, Fran Supek, Jenifer Muñoz, Laia Bonjoch, Anna Gratacós-Mulleras, Paula A. Sánchez-Rojas, Clara Esteban-Jurado, Teresa Ocaña, Miriam Cuatrecasas, Maria Vila-Casadesús, Juan José Lozano, Genis Parra, Steve Laurie, Sergi Beltran, EPICOLON Consortium, Antoni Castells, Luis Bujanda, Joaquín Cubiella, Francesc Balaguer and Sergi Castellví-Bel 100 80 10x ≥ age r 60 egions with cove r 40 ed r % of sha 20 0 I140 H458 H460 H461 H466 H468 H469 H470 FAM1 FAM3 FAM4 FAM19 FAM20 FAM22 FAM23 FAMN1 FAMN3 FAMN4 Families Figure S1. Histogram representing the percentage of genomic regions with a high-quality value of coverage (≥10×) with respect to all shared sequenced regions for each of the germline-tumor paired samples. Horizontal red line indicates sample filtering threshold (≥70% of shared regions with coverage above 10×). Figure S2. Pedigrees of the 18 families included in the study. Sample selected for germline and tumor whole-exome sequencing is indicated with an arrow. Filled symbols indicate affected for colorectal cancer (upper right quarter), adenoma/s (lower right quarter), gynecological cancer (ovary, uterine or breast cancer) (upper left quarter) and liver, stomach or pancreatic cancer (lower left quarter). Other cancer types are indicated in text with no symbol. IDs from samples undergoing germline whole-exome sequencing are also shown. AA/on-AA, advanced adenoma/non-advanced adenoma. Table S1. Description of germline copy number variants detected after calling with CoNIFER and ExomeDepth. -

RNA Editing at Baseline and Following Endoplasmic Reticulum Stress
RNA Editing at Baseline and Following Endoplasmic Reticulum Stress By Allison Leigh Richards A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Human Genetics) in The University of Michigan 2015 Doctoral Committee: Professor Vivian G. Cheung, Chair Assistant Professor Santhi K. Ganesh Professor David Ginsburg Professor Daniel J. Klionsky Dedication To my father, mother, and Matt without whom I would never have made it ii Acknowledgements Thank you first and foremost to my dissertation mentor, Dr. Vivian Cheung. I have learned so much from you over the past several years including presentation skills such as never sighing and never saying “as you can see…” You have taught me how to think outside the box and how to create and explain my story to others. I would not be where I am today without your help and guidance. Thank you to the members of my dissertation committee (Drs. Santhi Ganesh, David Ginsburg and Daniel Klionsky) for all of your advice and support. I would also like to thank the entire Human Genetics Program, and especially JoAnn Sekiguchi and Karen Grahl, for welcoming me to the University of Michigan and making my transition so much easier. Thank you to Michael Boehnke and the Genome Science Training Program for supporting my work. A very special thank you to all of the members of the Cheung lab, past and present. Thank you to Xiaorong Wang for all of your help from the bench to advice on my career. Thank you to Zhengwei Zhu who has helped me immensely throughout my thesis even through my panic. -

Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells
molecules Article Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells Michele Costanzo 1,2 , Marco Fiocchetti 3 , Paolo Ascenzi 3, Maria Marino 3 , Marianna Caterino 1,2,* and Margherita Ruoppolo 1,2,* 1 Department of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples Federico II, 80131 Naples, Italy; [email protected] 2 CEINGE—Biotecnologie Avanzate S.C.Ar.L., 80145 Naples, Italy 3 Department of Science, University Roma Tre, 00146 Rome, Italy; marco.fi[email protected] (M.F.); [email protected] (P.A.); [email protected] (M.M.) * Correspondence: [email protected] (M.C.); [email protected] (M.R.) Abstract: Neuroglobin (NGB) is a myoglobin-like monomeric globin that is involved in several processes, displaying a pivotal redox-dependent protective role in neuronal and extra-neuronal cells. NGB remarkably exerts its function upon upregulation by NGB inducers, such as 17β-estradiol (E2) and H2O2. However, the molecular bases of NGB’s functions remain undefined, mainly in non- neuronal cancer cells. Human MCF-7 breast cancer cells with a knocked-out (KO) NGB gene obtained using CRISPR/Cas9 technology were analyzed using shotgun label-free quantitative proteomics in comparison with control cells. The differential proteomics experiments were also performed after treatment with E2, H2O2, and E2 + H2O2. All the runs acquired using liquid chromatography–tandem mass spectrometry were elaborated within the same MaxQuant analysis, leading to the quantification Citation: Costanzo, M.; Fiocchetti, of 1872 proteins in the global proteomic dataset. Then, a differentially regulated protein dataset was M.; Ascenzi, P.; Marino, M.; Caterino, M.; Ruoppolo, M. -

Supplementary Table S1. Correlation Between the Mutant P53-Interacting Partners and PTTG3P, PTTG1 and PTTG2, Based on Data from Starbase V3.0 Database
Supplementary Table S1. Correlation between the mutant p53-interacting partners and PTTG3P, PTTG1 and PTTG2, based on data from StarBase v3.0 database. PTTG3P PTTG1 PTTG2 Gene ID Coefficient-R p-value Coefficient-R p-value Coefficient-R p-value NF-YA ENSG00000001167 −0.077 8.59e-2 −0.210 2.09e-6 −0.122 6.23e-3 NF-YB ENSG00000120837 0.176 7.12e-5 0.227 2.82e-7 0.094 3.59e-2 NF-YC ENSG00000066136 0.124 5.45e-3 0.124 5.40e-3 0.051 2.51e-1 Sp1 ENSG00000185591 −0.014 7.50e-1 −0.201 5.82e-6 −0.072 1.07e-1 Ets-1 ENSG00000134954 −0.096 3.14e-2 −0.257 4.83e-9 0.034 4.46e-1 VDR ENSG00000111424 −0.091 4.10e-2 −0.216 1.03e-6 0.014 7.48e-1 SREBP-2 ENSG00000198911 −0.064 1.53e-1 −0.147 9.27e-4 −0.073 1.01e-1 TopBP1 ENSG00000163781 0.067 1.36e-1 0.051 2.57e-1 −0.020 6.57e-1 Pin1 ENSG00000127445 0.250 1.40e-8 0.571 9.56e-45 0.187 2.52e-5 MRE11 ENSG00000020922 0.063 1.56e-1 −0.007 8.81e-1 −0.024 5.93e-1 PML ENSG00000140464 0.072 1.05e-1 0.217 9.36e-7 0.166 1.85e-4 p63 ENSG00000073282 −0.120 7.04e-3 −0.283 1.08e-10 −0.198 7.71e-6 p73 ENSG00000078900 0.104 2.03e-2 0.258 4.67e-9 0.097 3.02e-2 Supplementary Table S2. -

Cargo Specific Regulation of Cytoplasmic Dynein by Effector Proteins
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2018 Cargo Specific Regulation Of Cytoplasmic Dynein By Effector Proteins Mara Olenick University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, Biophysics Commons, and the Cell Biology Commons Recommended Citation Olenick, Mara, "Cargo Specific Regulation Of Cytoplasmic Dynein By Effector Proteins" (2018). Publicly Accessible Penn Dissertations. 3167. https://repository.upenn.edu/edissertations/3167 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3167 For more information, please contact [email protected]. Cargo Specific Regulation Of Cytoplasmic Dynein By Effector Proteins Abstract Axonal transport is vital for the development and survival of neurons. The transport of cargo and organelles from the axon to the cell body is driven almost completely by the molecular motor, cytoplasmic dynein. Yet, it remains unclear how dynein is spatially and temporally regulated given the variety of cargo that must be properly localized to maintain cellular function. Previous work has suggested that adaptor proteins provide a mechanism for cargo-specific egulationr of motors. During my thesis work, I have investigated the role of mammalian Hook proteins, Hook1 and Hook3, as potential motor adaptors. Using optogenetic and single molecule assays, I found that Hook proteins interact with both dynein and dynactin, to effectively activate dynein motility, inducing longer run lengths and higher velocities than the previously characterized dynein activator, BICD2. In addition, I found that complex formation requires the N-terminal domain of Hook proteins, which resembles the calponin-homology domain of EB proteins yet cannot bind directly to microtubules. -

Kif1b Rab7a Lmna
Title: Charcot-Marie-Tooth Neuropathy Type 2 GeneReview Molecular Genetics: Less Commonly Involved Genes Author: Bird TD Updated: March 2016 KIF1B Gene structure. KIF1B comprises 47 exons and 167.13 kb of DNA. Pathogenic allelic variants. See Table A, Locus Specific and HGMD Normal gene product. Kinesin-like protein KIF1B is involved in axonal transport of synaptic vesicle precursors [Zhao et al 2001]. The kinesin superfamily of proteins is essential for intracellular transport along microtubules. Abnormal gene product. There may be a defect in the transport of synaptic vesicles. RAB7A Gene structure. RAB7A has six exons and 87.9 kb of DNA. Pathogenic allelic variants. See Table A. Normal gene product. Ras-related protein Rab-7a belongs to the RAB family of Ras- related GTPases essential for the regulation of intracellular membrane trafficking. Rab- 7a is involved in transport between late endosomes and lysosomes. RAB-interacting lysosomal protein (RILP) induces the recruitment of dynein-dynactin motors and regulates transport toward the minus-end of microtubules [Verhoeven et al 2003]. Abnormal gene product. Abnormal Rab-7a may cause malfunction of lysosomes and inhibit neurite outgrowth [Spinosa et al 2008, Bucci & Deluca 2012]. LMNA Gene structure. LMNA has 12 exons spread over 24 kb of genomic DNA. Pathogenic allelic variants. The most common pathogenic variant found in individuals with CMT2B1 is p.Arg298Cys, a founder mutation in North Africa [Bouhouche et al 2007, De Sandre-Giovannoli et al 2002]. See also Table A. Table 5. Selected LMNA Variants DNA Nucleotide Protein Amino Acid Class of Variant Allele Reference Sequences Change Change Benign c.1908C>T p.= 1 c.398G>T p.Arg133Leu NM_170707.2 c.892C>T p.Arg298Cys Pathogenic NP_733821.1 c.1411C>T p.Arg471Cys c.1579C>T p.Arg527Cys Note on variant classification: Variants listed in the table have been provided by the author. -

Structural Characterization of the RH1-LZI Tandem of JIP3/4
www.nature.com/scientificreports OPEN Structural characterization of the RH1-LZI tandem of JIP3/4 highlights RH1 domains as a cytoskeletal motor-binding motif Fernando Vilela1, Christophe Velours1, Mélanie Chenon1, Magali Aumont-Nicaise1, Valérie Campanacci1, Aurélien Thureau2, Olena Pylypenko3, Jessica Andreani 1, Paola Llinas1* & Julie Ménétrey 1* JIP3 and JIP4 (JNK-interacting proteins 3 and 4) are adaptors for cargo recruitment by dynein/dynactin and kinesin1 motors. Both are dimers that are stabilised by two sections of leucine zipper coiled coils. The N-terminal Leucine Zipper I (LZI) belongs to a section that binds dynein-DLIC and kinesin1-KHC, whilst the medial Leucine Zipper II (LZII) binds dynactin-p150glued and kinesin1-KLC. Structural data is available for the LZII, but the LZI section is still uncharacterized. Here we characterize the N-terminal part of JIP3/4 which consists of an RH1 (RILP homology 1) domain followed by the LZI coiled coil using bioinformatical, biophysical and structural approaches. The RH1-LZI tandem of JIP3 associates as a high afnity homodimer exhibiting elongated alpha-helical fold. 3D homology modelling of the RH1-LZI tandem reveals that the kinesin1-KHC binding site mainly overlaps with the RH1 domain. A sequence comparison search indicates that only one other protein family has RH1 domains similar to those of JIP3/4, the RILP (Rab-interacting lysosomal protein) family which consists of adaptor proteins linking Rab GTPases to cytoskeletal motors. RILPL2 is recruited through its RH1 domain by the myosin 5a motor. Here, we showed that the RH1 domain of JIP3 also interacts with myosin 5 A in vitro, highlighting JIP3/4 as possible myosin 5a adaptors. -
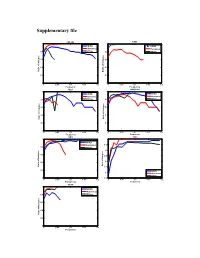
Supplementary File
Supplementary file COL1A2 FZD3 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of of indegree Ratio Ratio of Indegree 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency RBL1 SMARCA4 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree Ratio 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency SRC TP53 1 1 HPRD Humannet 0.95 0.8 PPIwu 0.9 e 0.6 0.85 0.8 0.4 Ratio of Indegree of Ratio Ratio of Indegre 0.75 0.2 HPRD 0.7 Humannet PPIwu 0 0.65 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency VCAN 1 HPRD Humannet 0.8 PPIwu 0.6 0.4 Ratio of Indegree 0.2 0 0 0.05 0.1 0.15 0.2 Frequency Figure S1. Variation of indegree ratio with frequency cutoffs from 0 to 0.2 with a step 0.002 for driver genes common by three fitness cores in COAD. The indegree ratio is set to NULL if total degree of this genes is less than 5. BRAF CASR 1 1 0.8 0.8 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree of Ratio HPRD HPRD 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency HDAC9 NRAS 1 1 0.8 0.8 0.6 0.6 0.4 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency NF1 CNTNAP2 1 1 0.9 0.8 0.8 0.6 0.7 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.6 Humannet 0.2 Humannet PPIwu PPIwu 0.5 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency Figure S2.