Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A Cause Malformations of Cortical Development and Microcephaly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genomic and Expression Profiling of Chromosome 17 in Breast Cancer Reveals Complex Patterns of Alterations and Novel Candidate Genes
[CANCER RESEARCH 64, 6453–6460, September 15, 2004] Genomic and Expression Profiling of Chromosome 17 in Breast Cancer Reveals Complex Patterns of Alterations and Novel Candidate Genes Be´atrice Orsetti,1 Me´lanie Nugoli,1 Nathalie Cervera,1 Laurence Lasorsa,1 Paul Chuchana,1 Lisa Ursule,1 Catherine Nguyen,2 Richard Redon,3 Stanislas du Manoir,3 Carmen Rodriguez,1 and Charles Theillet1 1Ge´notypes et Phe´notypes Tumoraux, EMI229 INSERM/Universite´ Montpellier I, Montpellier, France; 2ERM 206 INSERM/Universite´ Aix-Marseille 2, Parc Scientifique de Luminy, Marseille cedex, France; and 3IGBMC, U596 INSERM/Universite´Louis Pasteur, Parc d’Innovation, Illkirch cedex, France ABSTRACT 17q12-q21 corresponding to the amplification of ERBB2 and collinear genes, and a large region at 17q23 (5, 6). A number of new candidate Chromosome 17 is severely rearranged in breast cancer. Whereas the oncogenes have been identified, among which GRB7 and TOP2A at short arm undergoes frequent losses, the long arm harbors complex 17q21 or RP6SKB1, TBX2, PPM1D, and MUL at 17q23 have drawn combinations of gains and losses. In this work we present a comprehensive study of quantitative anomalies at chromosome 17 by genomic array- most attention (6–10). Furthermore, DNA microarray studies have comparative genomic hybridization and of associated RNA expression revealed additional candidates, with some located outside current changes by cDNA arrays. We built a genomic array covering the entire regions of gains, thus suggesting the existence of additional amplicons chromosome at an average density of 1 clone per 0.5 Mb, and patterns of on 17q (8, 9). gains and losses were characterized in 30 breast cancer cell lines and 22 Our previous loss of heterozygosity mapping data pointed to the primary tumors. -

Actin Nucleator Spire 1 Is a Regulator of Ectoplasmic Specialization in the Testis Qing Wen1,Nanli1,Xiangxiao 1,2,Wing-Yeelui3, Darren S
Wen et al. Cell Death and Disease (2018) 9:208 DOI 10.1038/s41419-017-0201-6 Cell Death & Disease ARTICLE Open Access Actin nucleator Spire 1 is a regulator of ectoplasmic specialization in the testis Qing Wen1,NanLi1,XiangXiao 1,2,Wing-yeeLui3, Darren S. Chu1, Chris K. C. Wong4, Qingquan Lian5,RenshanGe5, Will M. Lee3, Bruno Silvestrini6 and C. Yan Cheng 1 Abstract Germ cell differentiation during the epithelial cycle of spermatogenesis is accompanied by extensive remodeling at the Sertoli cell–cell and Sertoli cell–spermatid interface to accommodate the transport of preleptotene spermatocytes and developing spermatids across the blood–testis barrier (BTB) and the adluminal compartment of the seminiferous epithelium, respectively. The unique cell junction in the testis is the actin-rich ectoplasmic specialization (ES) designated basal ES at the Sertoli cell–cell interface, and the apical ES at the Sertoli–spermatid interface. Since ES dynamics (i.e., disassembly, reassembly and stabilization) are supported by actin microfilaments, which rapidly converts between their bundled and unbundled/branched configuration to confer plasticity to the ES, it is logical to speculate that actin nucleation proteins play a crucial role to ES dynamics. Herein, we reported findings that Spire 1, an actin nucleator known to polymerize actins into long stretches of linear microfilaments in cells, is an important regulator of ES dynamics. Its knockdown by RNAi in Sertoli cells cultured in vitro was found to impede the Sertoli cell tight junction (TJ)-permeability barrier through changes in the organization of F-actin across Sertoli cell cytosol. Unexpectedly, Spire 1 knockdown also perturbed microtubule (MT) organization in Sertoli cells cultured in vitro. -

Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells
molecules Article Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells Michele Costanzo 1,2 , Marco Fiocchetti 3 , Paolo Ascenzi 3, Maria Marino 3 , Marianna Caterino 1,2,* and Margherita Ruoppolo 1,2,* 1 Department of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples Federico II, 80131 Naples, Italy; [email protected] 2 CEINGE—Biotecnologie Avanzate S.C.Ar.L., 80145 Naples, Italy 3 Department of Science, University Roma Tre, 00146 Rome, Italy; marco.fi[email protected] (M.F.); [email protected] (P.A.); [email protected] (M.M.) * Correspondence: [email protected] (M.C.); [email protected] (M.R.) Abstract: Neuroglobin (NGB) is a myoglobin-like monomeric globin that is involved in several processes, displaying a pivotal redox-dependent protective role in neuronal and extra-neuronal cells. NGB remarkably exerts its function upon upregulation by NGB inducers, such as 17β-estradiol (E2) and H2O2. However, the molecular bases of NGB’s functions remain undefined, mainly in non- neuronal cancer cells. Human MCF-7 breast cancer cells with a knocked-out (KO) NGB gene obtained using CRISPR/Cas9 technology were analyzed using shotgun label-free quantitative proteomics in comparison with control cells. The differential proteomics experiments were also performed after treatment with E2, H2O2, and E2 + H2O2. All the runs acquired using liquid chromatography–tandem mass spectrometry were elaborated within the same MaxQuant analysis, leading to the quantification Citation: Costanzo, M.; Fiocchetti, of 1872 proteins in the global proteomic dataset. Then, a differentially regulated protein dataset was M.; Ascenzi, P.; Marino, M.; Caterino, M.; Ruoppolo, M. -

Gamma Tubulin (TUBG1) (NM 001070) Human Untagged Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC119462 gamma Tubulin (TUBG1) (NM_001070) Human Untagged Clone Product data: Product Type: Expression Plasmids Product Name: gamma Tubulin (TUBG1) (NM_001070) Human Untagged Clone Tag: Tag Free Symbol: TUBG1 Synonyms: CDCBM4; GCP-1; TUBG; TUBGCP1 Vector: pCMV6-XL5 E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: None Fully Sequenced ORF: >OriGene ORF within SC119462 sequence for NM_001070 edited (data generated by NextGen Sequencing) ATGCCGAGGGAAATCATCACCCTACAGTTGGGCCAGTGCGGCAATCAGATTGGGTTCGAG TTCTGGAAACAGCTGTGCGCCGAGCATGGTATCAGCCCCGAGGGCATCGTGGAGGAGTTC GCCACCGAGGGCACTGACCGCAAGGACGTCTTTTTCTACCAGGCAGACGATGAGCACTAC ATCCCCCGGGCCGTGCTGCTGGACTTGGAACCCCGGGTGATCCACTCCATCCTCAACTCC CCCTATGCCAAGCTCTACAACCCAGAGAACATCTACCTGTCGGAACATGGAGGAGGAGCT GGCAACAACTGGGCCAGCGGATTCTCCCAGGGAGAAAAGATCCATGAGGACATTTTTGAC ATCATAGACCGGGAGGCAGATGGTAGTGACAGTCTAGAGGGCTTTGTGCTGTGTCACTCC ATTGCTGGGGGGACAGGCTCTGGACTGGGTTCCTACCTCTTAGAACGGCTGAATGACAGG TATCCTAAGAAGCTGGTGCAGACATACTCAGTGTTTCCCAACCAGGACGAGATGAGCGAT GTGGTGGTCCAGCCTTACAATTCACTCCTCACACTCAAGAGGCTGACGCAGAATGCAGAC TGTGTGGTGGTGCTGGACAACACAGCCCTGAACCGGATTGCCACAGACCGCCTGCACATC CAGAACCCATCCTTCTCCCAGATCAACCAGCTGGTGTCTACCATCATGTCAGCCAGCACC ACCACCCTGCGCTACCCTGGCTACATGAACAATGACCTCATCGGCCTCATCGCCTCGCTC ATTCCCACCCCACGGCTCCACTTCCTCATGACCGGCTACACCCCTCTCACTACGGACCAG TCAGTGGCCAGCGTGAGGAAGACCACGGTCCTGGATGTCATGAGGCGGCTGCTGCAGCCC -

Supplementary Table S1. Correlation Between the Mutant P53-Interacting Partners and PTTG3P, PTTG1 and PTTG2, Based on Data from Starbase V3.0 Database
Supplementary Table S1. Correlation between the mutant p53-interacting partners and PTTG3P, PTTG1 and PTTG2, based on data from StarBase v3.0 database. PTTG3P PTTG1 PTTG2 Gene ID Coefficient-R p-value Coefficient-R p-value Coefficient-R p-value NF-YA ENSG00000001167 −0.077 8.59e-2 −0.210 2.09e-6 −0.122 6.23e-3 NF-YB ENSG00000120837 0.176 7.12e-5 0.227 2.82e-7 0.094 3.59e-2 NF-YC ENSG00000066136 0.124 5.45e-3 0.124 5.40e-3 0.051 2.51e-1 Sp1 ENSG00000185591 −0.014 7.50e-1 −0.201 5.82e-6 −0.072 1.07e-1 Ets-1 ENSG00000134954 −0.096 3.14e-2 −0.257 4.83e-9 0.034 4.46e-1 VDR ENSG00000111424 −0.091 4.10e-2 −0.216 1.03e-6 0.014 7.48e-1 SREBP-2 ENSG00000198911 −0.064 1.53e-1 −0.147 9.27e-4 −0.073 1.01e-1 TopBP1 ENSG00000163781 0.067 1.36e-1 0.051 2.57e-1 −0.020 6.57e-1 Pin1 ENSG00000127445 0.250 1.40e-8 0.571 9.56e-45 0.187 2.52e-5 MRE11 ENSG00000020922 0.063 1.56e-1 −0.007 8.81e-1 −0.024 5.93e-1 PML ENSG00000140464 0.072 1.05e-1 0.217 9.36e-7 0.166 1.85e-4 p63 ENSG00000073282 −0.120 7.04e-3 −0.283 1.08e-10 −0.198 7.71e-6 p73 ENSG00000078900 0.104 2.03e-2 0.258 4.67e-9 0.097 3.02e-2 Supplementary Table S2. -

Applying Expression Profile Similarity for Discovery of Patient-Specific
bioRxiv preprint doi: https://doi.org/10.1101/172015; this version posted September 17, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Applying expression profile similarity for discovery of patient-specific functional mutations Guofeng Meng Partner Institute of Computational Biology, Yueyang 333, Shanghai, China email: [email protected] Abstract The progress of cancer genome sequencing projects yields unprecedented information of mutations for numerous patients. However, the complexity of mutation profiles of patients hinders the further understanding of mechanisms of oncogenesis. One basic question is how to uncover mutations with functional impacts. In this work, we introduce a computational method to predict functional somatic mutations for each of patient by integrating mutation recurrence with similarity of expression profiles of patients. With this method, the functional mutations are determined by checking the mutation enrichment among a group of patients with similar expression profiles. We applied this method to three cancer types and identified the functional mutations. Comparison of the predictions for three cancer types suggested that most of the functional mutations were cancer-type-specific with one exception to p53. By checking prediction results, we found that our method effectively filtered non-functional mutations resulting from large protein sizes. In addition, this methods can also perform functional annotation to each patient to describe their association with signalling pathways or biological processes. In breast cancer, we predicted "cell adhesion" and other mutated gene associated terms to be significantly enriched among patients. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Involvement of the Kinesin Family Members KIF4A and KIF5C In
Downloaded from http://jmg.bmj.com/ on March 10, 2015 - Published by group.bmj.com Cognitive and behavioural genetics ORIGINAL ARTICLE Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic Editor’s choice Scan to access more free content function Marjolein H Willemsen,1,2 Wei Ba,1,3,4 Willemijn M Wissink-Lindhout,1 Arjan P M de Brouwer,1,2 Stefan A Haas,5 Melanie Bienek,6 Hao Hu,6 Lisenka E L M Vissers,1,2 Hans van Bokhoven,1,2,3,4 Vera Kalscheuer,6 Nael Nadif Kasri,1,2,3,4 Tjitske Kleefstra1,2 For numbered affiliations see ABSTRACT genes in the development and functioning of the end of article. Introduction Kinesin superfamily (KIF) genes encode nervous system. Mice with homozygous knockout Kif1a 1b 2a 3a 3b 4a 5a 5b Correspondence to motor proteins that have fundamental roles in brain mutations in , , , , , , and Dr Nael Nadif Kasri, functioning, development, survival and plasticity by show various neurological phenotypes including Department of Cognitive regulating the transport of cargo along microtubules structural brain anomalies, decreased brain size, Neuroscience, Radboud within axons, dendrites and synapses. Mouse knockout loss of neurons, reduced rate of neuronal apoptosis university medical center, studies support these important functions in the nervous and perinatal lethality due to neurological pro- Nijmegen, The Netherlands; 4–12 Nael.NadifKasri@radboudumc. system. The role of KIF genes in intellectual disability (ID) blems. The embryonic lethality of knockout nl; has so far received limited attention, although previous mice for Kif2a, Kif3a and 3b, and Kif5b suggest Dr Tjitske Kleefstra, studies have suggested that many ID genes impinge on that these Kif genes have an important function in Department of Human synaptic function. -

Kif1b Rab7a Lmna
Title: Charcot-Marie-Tooth Neuropathy Type 2 GeneReview Molecular Genetics: Less Commonly Involved Genes Author: Bird TD Updated: March 2016 KIF1B Gene structure. KIF1B comprises 47 exons and 167.13 kb of DNA. Pathogenic allelic variants. See Table A, Locus Specific and HGMD Normal gene product. Kinesin-like protein KIF1B is involved in axonal transport of synaptic vesicle precursors [Zhao et al 2001]. The kinesin superfamily of proteins is essential for intracellular transport along microtubules. Abnormal gene product. There may be a defect in the transport of synaptic vesicles. RAB7A Gene structure. RAB7A has six exons and 87.9 kb of DNA. Pathogenic allelic variants. See Table A. Normal gene product. Ras-related protein Rab-7a belongs to the RAB family of Ras- related GTPases essential for the regulation of intracellular membrane trafficking. Rab- 7a is involved in transport between late endosomes and lysosomes. RAB-interacting lysosomal protein (RILP) induces the recruitment of dynein-dynactin motors and regulates transport toward the minus-end of microtubules [Verhoeven et al 2003]. Abnormal gene product. Abnormal Rab-7a may cause malfunction of lysosomes and inhibit neurite outgrowth [Spinosa et al 2008, Bucci & Deluca 2012]. LMNA Gene structure. LMNA has 12 exons spread over 24 kb of genomic DNA. Pathogenic allelic variants. The most common pathogenic variant found in individuals with CMT2B1 is p.Arg298Cys, a founder mutation in North Africa [Bouhouche et al 2007, De Sandre-Giovannoli et al 2002]. See also Table A. Table 5. Selected LMNA Variants DNA Nucleotide Protein Amino Acid Class of Variant Allele Reference Sequences Change Change Benign c.1908C>T p.= 1 c.398G>T p.Arg133Leu NM_170707.2 c.892C>T p.Arg298Cys Pathogenic NP_733821.1 c.1411C>T p.Arg471Cys c.1579C>T p.Arg527Cys Note on variant classification: Variants listed in the table have been provided by the author. -
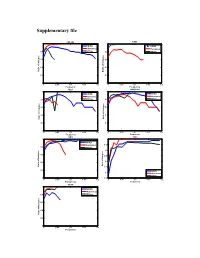
Supplementary File
Supplementary file COL1A2 FZD3 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of of indegree Ratio Ratio of Indegree 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency RBL1 SMARCA4 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree Ratio 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency SRC TP53 1 1 HPRD Humannet 0.95 0.8 PPIwu 0.9 e 0.6 0.85 0.8 0.4 Ratio of Indegree of Ratio Ratio of Indegre 0.75 0.2 HPRD 0.7 Humannet PPIwu 0 0.65 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency VCAN 1 HPRD Humannet 0.8 PPIwu 0.6 0.4 Ratio of Indegree 0.2 0 0 0.05 0.1 0.15 0.2 Frequency Figure S1. Variation of indegree ratio with frequency cutoffs from 0 to 0.2 with a step 0.002 for driver genes common by three fitness cores in COAD. The indegree ratio is set to NULL if total degree of this genes is less than 5. BRAF CASR 1 1 0.8 0.8 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree of Ratio HPRD HPRD 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency HDAC9 NRAS 1 1 0.8 0.8 0.6 0.6 0.4 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency NF1 CNTNAP2 1 1 0.9 0.8 0.8 0.6 0.7 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.6 Humannet 0.2 Humannet PPIwu PPIwu 0.5 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency Figure S2. -

A Master Autoantigen-Ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A Master Autoantigen-ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases Julia Y. Wang1*, Michael W. Roehrl1, Victor B. Roehrl1, and Michael H. Roehrl2* 1 Curandis, New York, USA 2 Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA * Correspondence: [email protected] or [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Chronic and debilitating autoimmune sequelae pose a grave concern for the post-COVID-19 pandemic era. Based on our discovery that the glycosaminoglycan dermatan sulfate (DS) displays peculiar affinity to apoptotic cells and autoantigens (autoAgs) and that DS-autoAg complexes cooperatively stimulate autoreactive B1 cell responses, we compiled a database of 751 candidate autoAgs from six human cell types. At least 657 of these have been found to be affected by SARS-CoV-2 infection based on currently available multi-omic COVID data, and at least 400 are confirmed targets of autoantibodies in a wide array of autoimmune diseases and cancer.