Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neurogenic Decisions Require a Cell Cycle Independent Function of The
RESEARCH ARTICLE Neurogenic decisions require a cell cycle independent function of the CDC25B phosphatase Fre´ de´ ric Bonnet1†, Angie Molina1†, Me´ lanie Roussat1, Manon Azais2, Sophie Bel-Vialar1, Jacques Gautrais2, Fabienne Pituello1*, Eric Agius1* 1Centre de Biologie du De´veloppement, Centre de Biologie Inte´grative, Universite´ de Toulouse, CNRS, UPS, Toulouse, France; 2Centre de Recherches sur la Cognition Animale, Centre de Biologie Inte´grative., Universite´ de Toulouse, CNRS, UPS, Toulouse, France Abstract A fundamental issue in developmental biology and in organ homeostasis is understanding the molecular mechanisms governing the balance between stem cell maintenance and differentiation into a specific lineage. Accumulating data suggest that cell cycle dynamics play a major role in the regulation of this balance. Here we show that the G2/M cell cycle regulator CDC25B phosphatase is required in mammals to finely tune neuronal production in the neural tube. We show that in chick neural progenitors, CDC25B activity favors fast nuclei departure from the apical surface in early G1, stimulates neurogenic divisions and promotes neuronal differentiation. We design a mathematical model showing that within a limited period of time, cell cycle length modifications cannot account for changes in the ratio of the mode of division. Using a CDC25B point mutation that cannot interact with CDK, we show that part of CDC25B activity is independent *For correspondence: of its action on the cell cycle. [email protected] (FP); [email protected] (EA) †These authors contributed equally to this work Introduction In multicellular organisms, managing the development, homeostasis and regeneration of tissues Competing interests: The requires the tight control of self-renewal and differentiation of stem/progenitor cells. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

On Cellular Automaton Approaches to Modeling Biological Cells
ON CELLULAR AUTOMATON APPROACHES TO MODELING BIOLOGICAL CELLS MARK S. ALBER∗, MARIA A. KISKOWSKIy , JAMES A. GLAZIERz , AND YI JIANGx Abstract. We discuss two different types of Cellular Automata (CA): lattice-gas- based cellular automata (LGCA) and the cellular Potts model (CPM), and describe their applications in biological modeling. LGCA were originally developed for modeling ideal gases and fluids. We describe several extensions of the classical LGCA model to self-driven biological cells. In partic- ular, we review recent models for rippling in myxobacteria, cell aggregation, swarming, and limb bud formation. These LGCA-based models show the versatility of CA in modeling and their utility in addressing basic biological questions. The CPM is a more sophisticated CA, which describes individual cells as extended objects of variable shape. We review various extensions to the original Potts model and describe their application to morphogenesis; the development of a complex spatial struc- ture by a collection of cells. We focus on three phenomena: cell sorting in aggregates of embryonic chicken cells, morphological development of the slime mold Dictyostelium discoideum and avascular tumor growth. These models include intercellular and extra- cellular interactions, as well as cell growth and death. 1. Introduction. Cellular automata (CA) consist of discrete agents or particles, which occupy some or all sites of a regular lattice. These par- ticles have one or more internal state variables (which may be discrete or continuous) and a set of rules describing the evolution of their state and position (in older models, particles usually occupied all lattice sites, one particle per node, and did not move). -

Supplementary Table S1. Correlation Between the Mutant P53-Interacting Partners and PTTG3P, PTTG1 and PTTG2, Based on Data from Starbase V3.0 Database
Supplementary Table S1. Correlation between the mutant p53-interacting partners and PTTG3P, PTTG1 and PTTG2, based on data from StarBase v3.0 database. PTTG3P PTTG1 PTTG2 Gene ID Coefficient-R p-value Coefficient-R p-value Coefficient-R p-value NF-YA ENSG00000001167 −0.077 8.59e-2 −0.210 2.09e-6 −0.122 6.23e-3 NF-YB ENSG00000120837 0.176 7.12e-5 0.227 2.82e-7 0.094 3.59e-2 NF-YC ENSG00000066136 0.124 5.45e-3 0.124 5.40e-3 0.051 2.51e-1 Sp1 ENSG00000185591 −0.014 7.50e-1 −0.201 5.82e-6 −0.072 1.07e-1 Ets-1 ENSG00000134954 −0.096 3.14e-2 −0.257 4.83e-9 0.034 4.46e-1 VDR ENSG00000111424 −0.091 4.10e-2 −0.216 1.03e-6 0.014 7.48e-1 SREBP-2 ENSG00000198911 −0.064 1.53e-1 −0.147 9.27e-4 −0.073 1.01e-1 TopBP1 ENSG00000163781 0.067 1.36e-1 0.051 2.57e-1 −0.020 6.57e-1 Pin1 ENSG00000127445 0.250 1.40e-8 0.571 9.56e-45 0.187 2.52e-5 MRE11 ENSG00000020922 0.063 1.56e-1 −0.007 8.81e-1 −0.024 5.93e-1 PML ENSG00000140464 0.072 1.05e-1 0.217 9.36e-7 0.166 1.85e-4 p63 ENSG00000073282 −0.120 7.04e-3 −0.283 1.08e-10 −0.198 7.71e-6 p73 ENSG00000078900 0.104 2.03e-2 0.258 4.67e-9 0.097 3.02e-2 Supplementary Table S2. -

Increased Processing of SINE B2 Ncrnas Unveils a Novel Type Of
RESEARCH ARTICLE Increased processing of SINE B2 ncRNAs unveils a novel type of transcriptome deregulation in amyloid beta neuropathology Yubo Cheng1,2,3,4†, Luke Saville1,2,3,4†, Babita Gollen1,2,3,4†, Christopher Isaac1,2,3,4†, Abel Belay1,2,3,4, Jogender Mehla3, Kush Patel1,2,3, Nehal Thakor1,2,3, Majid H Mohajerani3, Athanasios Zovoilis1,2,3,4* 1Department of Chemistry and Biochemistry, University of Lethbridge, Lethbridge, Canada; 2Southern Alberta Genome Sciences Centre, University of Lethbridge, Lethbridge, Canada; 3Canadian Centre for Behavioral Neuroscience, University of Lethbridge, Lethbridge, Canada; 4Alberta RNA Research and Training Institute, University of Lethbridge, Lethbridge, Canada Abstract The functional importance of many non-coding RNAs (ncRNAs) generated by repetitive elements and their connection with pathologic processes remains elusive. B2 RNAs, a class of ncRNAs of the B2 family of SINE repeats, mediate through their processing the transcriptional activation of various genes in response to stress. Here, we show that this response is dysfunctional during amyloid beta toxicity and pathology in the mouse hippocampus due to increased levels of B2 RNA processing, leading to constitutively elevated B2 RNA target gene expression and high Trp53 levels. Evidence indicates that Hsf1, a master regulator of stress response, mediates B2 RNA *For correspondence: processing in hippocampal cells and is activated during amyloid toxicity, accelerating the [email protected] processing of SINE RNAs and gene hyper-activation. Our study reveals that in mouse, SINE RNAs † constitute a novel pathway deregulated in amyloid beta pathology, with potential implications for These authors contributed equally to this work similar cases in the human brain, such as Alzheimer’s disease (AD). -

Kif1b Rab7a Lmna
Title: Charcot-Marie-Tooth Neuropathy Type 2 GeneReview Molecular Genetics: Less Commonly Involved Genes Author: Bird TD Updated: March 2016 KIF1B Gene structure. KIF1B comprises 47 exons and 167.13 kb of DNA. Pathogenic allelic variants. See Table A, Locus Specific and HGMD Normal gene product. Kinesin-like protein KIF1B is involved in axonal transport of synaptic vesicle precursors [Zhao et al 2001]. The kinesin superfamily of proteins is essential for intracellular transport along microtubules. Abnormal gene product. There may be a defect in the transport of synaptic vesicles. RAB7A Gene structure. RAB7A has six exons and 87.9 kb of DNA. Pathogenic allelic variants. See Table A. Normal gene product. Ras-related protein Rab-7a belongs to the RAB family of Ras- related GTPases essential for the regulation of intracellular membrane trafficking. Rab- 7a is involved in transport between late endosomes and lysosomes. RAB-interacting lysosomal protein (RILP) induces the recruitment of dynein-dynactin motors and regulates transport toward the minus-end of microtubules [Verhoeven et al 2003]. Abnormal gene product. Abnormal Rab-7a may cause malfunction of lysosomes and inhibit neurite outgrowth [Spinosa et al 2008, Bucci & Deluca 2012]. LMNA Gene structure. LMNA has 12 exons spread over 24 kb of genomic DNA. Pathogenic allelic variants. The most common pathogenic variant found in individuals with CMT2B1 is p.Arg298Cys, a founder mutation in North Africa [Bouhouche et al 2007, De Sandre-Giovannoli et al 2002]. See also Table A. Table 5. Selected LMNA Variants DNA Nucleotide Protein Amino Acid Class of Variant Allele Reference Sequences Change Change Benign c.1908C>T p.= 1 c.398G>T p.Arg133Leu NM_170707.2 c.892C>T p.Arg298Cys Pathogenic NP_733821.1 c.1411C>T p.Arg471Cys c.1579C>T p.Arg527Cys Note on variant classification: Variants listed in the table have been provided by the author. -

Genetic Algorithms and Their Application to In-Silico Evolution of Genetic Regulatory Networks
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Hertfordshire Research Archive Genetic Algorithms and their Application to in-silico Evolution of Genetic Regulatory Networks Johannes F. Knabe‡†, Katja Wegner†, Chrystopher L. Nehaniv‡†, and Maria J. Schilstra†* †Biological and Neural Computation Laboratory, and ‡Adaptive Systems Research Group, STRI, University of Hertfordshire, Hatfield, AL10 9AB United Kingdom *To whom correspondence should be addressed ([email protected]) i. Abstract A genetic algorithm (GA) is a procedure that mimics processes occurring in Darwinian evolution to solve computational problems. A GA introduces variation through “mutation” and “recombination” in a “population” of possible solutions to a problem, encoded as strings of characters in “genomes”, and allows this population to evolve, using selection procedures that favor the gradual enrichment of the gene pool with the genomes of the “fitter” individuals. GA’s are particularly suitable for optimization problems in which an effective system design or set of parameter values is sought. In nature, genetic regulatory networks (GRN’s) form the basic control layer in the regulation of gene expression levels. GRN’s are composed of regulatory interactions between genes and their gene products, and are, inter alia, at the basis of the development of single fertilized cells into fully grown organisms. This paper describes how GA’s may be applied to find functional regulatory schemes and parameter values for models that capture the fundamental GRN characteristics. The central ideas behind evolutionary computation and GRN modeling, and the considerations in GA design and use are discussed, and illustrated with an extended example. -
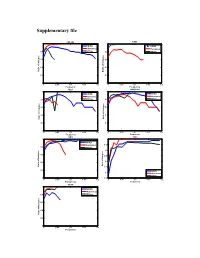
Supplementary File
Supplementary file COL1A2 FZD3 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of of indegree Ratio Ratio of Indegree 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency RBL1 SMARCA4 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree Ratio 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency SRC TP53 1 1 HPRD Humannet 0.95 0.8 PPIwu 0.9 e 0.6 0.85 0.8 0.4 Ratio of Indegree of Ratio Ratio of Indegre 0.75 0.2 HPRD 0.7 Humannet PPIwu 0 0.65 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency VCAN 1 HPRD Humannet 0.8 PPIwu 0.6 0.4 Ratio of Indegree 0.2 0 0 0.05 0.1 0.15 0.2 Frequency Figure S1. Variation of indegree ratio with frequency cutoffs from 0 to 0.2 with a step 0.002 for driver genes common by three fitness cores in COAD. The indegree ratio is set to NULL if total degree of this genes is less than 5. BRAF CASR 1 1 0.8 0.8 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree of Ratio HPRD HPRD 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency HDAC9 NRAS 1 1 0.8 0.8 0.6 0.6 0.4 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency NF1 CNTNAP2 1 1 0.9 0.8 0.8 0.6 0.7 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.6 Humannet 0.2 Humannet PPIwu PPIwu 0.5 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency Figure S2. -

Self-Reproduction Based on Turing Machines
MEASURES OF ORGANIZATION IN A MODEL OF CELLULAR SELF-REPRODUCTION BASED ON TURING MACHINES WALTER R. STAHL OREGON REGIONAL PRIMATE RESEARCH CENTER BEAVERTON, OREGON 1. Introductory summary The late John von Neumann first realized that self-reproduction, as observed in biological systems, was a legitimate problem of mathematics, specifically of automata and algorithm theory. He founded the study of self-reproduction in systems of "natural automata." This report deals with the self-organizing prop- erties of a new type of cellular self-reproduction model. The action of enzymes is simulated by Turing machines acting as molecular automata or computers, with their highly standardized coding corresponding to genes of the cell. Com- puter experiments have been done to test the stability and logical homeostatic properties of a 36 gene cell model. It is shown that the system will continue to organize itself and reproduce in spite of a variety of environmental deficiencies and disturbances, and also to repair itself after certain kinds of injury. The most pertinent organizational criteria for the described model are amounts of "energy" and "time cycles" needed to reach maturity and reproduce. Comparisons are drawn between these organizational measures and thermodynamic informa- tional parameters such as Gibbsean chemical potential and entropy. The total amount of genetic and cellular information can be quantified in the cell model and this helps clarify some confusing aspects of genetic information measures. The model is highly idealized. It bears somewhat the same relationship to real cells as computer circuits do to the brain, but shows that automata theory can be applied to molecular biology in a meaningful way. -

A Master Autoantigen-Ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A Master Autoantigen-ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases Julia Y. Wang1*, Michael W. Roehrl1, Victor B. Roehrl1, and Michael H. Roehrl2* 1 Curandis, New York, USA 2 Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA * Correspondence: [email protected] or [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Chronic and debilitating autoimmune sequelae pose a grave concern for the post-COVID-19 pandemic era. Based on our discovery that the glycosaminoglycan dermatan sulfate (DS) displays peculiar affinity to apoptotic cells and autoantigens (autoAgs) and that DS-autoAg complexes cooperatively stimulate autoreactive B1 cell responses, we compiled a database of 751 candidate autoAgs from six human cell types. At least 657 of these have been found to be affected by SARS-CoV-2 infection based on currently available multi-omic COVID data, and at least 400 are confirmed targets of autoantibodies in a wide array of autoimmune diseases and cancer. -

Metabolic Network Modeling with Model Organisms
Available online at www.sciencedirect.com ScienceDirect Metabolic network modeling with model organisms L Safak Yilmaz and Albertha JM Walhout Flux balance analysis (FBA) with genome-scale metabolic reconstruct a genome-scale metabolic network model network models (GSMNM) allows systems level predictions of (GSMNM), which is then used to calculate the flux metabolism in a variety of organisms. Different types of distribution over the entire network in any defined con- predictions with different accuracy levels can be made dition for the organism (see below). For model organisms, depending on the applied experimental constraints ranging high-quality genomic annotations allow the reconstruc- from measurement of exchange fluxes to the integration of tion of comprehensive GSMNMs that can be parame- gene expression data. Metabolic network modeling with model terized and validated with publically available experi- organisms has pioneered method development in this field. In mental datasets. In addition, since many properties of addition, model organism GSMNMs are useful for basic metabolic networks are conserved across taxa, model understanding of metabolism, and in the case of animal organism GSMNMs can be used to study human disease models, for the study of metabolic human diseases. Here, we with animal models, while plant models can instruct discuss GSMNMs of most highly used model organisms with agriculture. the emphasis on recent reconstructions. Here, we first summarize the basics of genome-scale meta- Address bolic network modeling, and then explore GSMNMs Program in Systems Biology, Program in Molecular Medicine, University and their applications in common model organisms [3] of Massachusetts Medical School, Worcester, MA, United States (Figure 1). -

The Unfolded Protein Response and Its Potential Role In
F1000Research 2016, 4:103 Last updated: 22 JUL 2020 RESEARCH ARTICLE The unfolded protein response and its potential role in Huntington's disease elucidated by a systems biology approach [version 2; peer review: 2 approved] Ravi Kiran Reddy Kalathur1, Joaquin Giner-Lamia1, Susana Machado1, Tania Barata1, Kameshwar R S Ayasolla1, Matthias E. Futschik1,2 1Centre for Biomedical Research, University of Algarve, Faro, 8005-139, Portugal 2Centre of Marine Sciences, University of Algarve, Faro, 8005-139, Portugal First published: 01 May 2015, 4:103 Open Peer Review v2 https://doi.org/10.12688/f1000research.6358.1 Latest published: 02 Mar 2016, 4:103 https://doi.org/10.12688/f1000research.6358.2 Reviewer Status Abstract Invited Reviewers Huntington ́s disease (HD) is a progressive, neurodegenerative disease 1 2 with a fatal outcome. Although the disease-causing gene (huntingtin) has been known for over 20 years, the exact mechanisms leading to neuronal version 2 cell death are still controversial. One potential mechanism contributing to (revision) report the massive loss of neurons observed in the brain of HD patients could be 02 Mar 2016 the unfolded protein response (UPR) activated by accumulation of misfolded proteins in the endoplasmic reticulum (ER). As an adaptive response to counter-balance accumulation of un- or misfolded proteins, the version 1 UPR upregulates transcription of chaperones, temporarily attenuates new 01 May 2015 report report translation, and activates protein degradation via the proteasome. However, persistent ER stress and an activated UPR can also cause apoptotic cell death. Although different studies have indicated a role for the Scott Zeitlin, University of Virginia, UPR in HD, the evidence remains inconclusive.