Renal Coloboma Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Genetic and Developmental Basis of Renal Coloboma (Papillorenal) Syndrome
Perspective Genetic and developmental basis of renal coloboma (papillorenal) syndrome Expert Rev. Ophthalmol. 4(2), 135–xxx (2009) Lisa A Schimmenti Renal coloboma syndrome, also known as papillorenal syndrome, is characterized by optic Department of Pediatrics and nerve anomalies and kidney hypodysplasia. Autosomal dominant mutations in the gene Ophthalmology, University of encoding the paired box transcription factor PAX2 can be identified in nearly half of all Minnesota Medical School, 420 patients with this phenotype. The primary ophthalmologic findings include congenital central Delaware Street Southeast, retinal vasculature absence associated with abnormalities in retinal blood vessel patterning MMC 730, Minnesota, and deeply excavated optic discs. Other published findings include optic nerve hypoplasia, MN 55455, USA optic nerve cyst, optic nerve pits, retinal coloboma, microphthalmia and scleral staphyloma. Tel.: +1 612 624 5613 Visual acuity ranges from normal to severe impairment. Up to one third of affected patients Fax: +1 612 626 2993 will develop end-stage renal disease. Mouse and zebrafish withPax2/pax2a mutations provide [email protected] developmentally based explanations for the observed phenotypic observations in affected patients. KEYWORDS: optic nerve coloboma • optic nerve dysplasia • papillorenal syndrome • PAX2 • renal coloboma syndrome The clinical presentation of optic nerve anomalies total of 10 years later, Weaver et al. reported a case associated with renal hypodysplasia should alert the of two brothers, both with optic nerve colobomas, clinician to the possibility that a patient may have interstitial nephritis and renal failure [3]. Weaver renal coloboma syndrome, a condition also known noted the similarity of the optic nerve colobomas as papillorenal syndrome (OMIM#120330). The in his patients with the ‘morning glory anomaly’, optic nerve findings could be described as a ‘dys- previously reported by Karcher et al. -

Ophthalmology
Ophthalmology Information for health professionals MEDICAL GENETIC TESTING FOR OPHTHALMOLOGY Recent technologies, in particularly Next Generation Sequencing (NGS), allows fast, accurate and valuable diagnostic tests. For Ophthalmology, CGC Genetics has an extensive list of medical genetic tests with clinical integration of results by our Medical Geneticists. 1. EXOME SEQUENCING: Exome Sequencing is a very efficient strategy to study most exons of a patient’s genome, unraveling mutations associated with specific disorders or phenotypes. With this diagnostic strategy, patients can be studied with a significantly reduced turnaround time and cost. CGC Genetics has available 2 options for Exome Sequencing: • Whole Exome Sequencing (WES), which analyzes the entire exome (about 20 000 genes); • Disease Exome by CGC Genetics, which analyzes about 6 000 clinically-relevant genes. Any of these can be performed in the index case or in a Trio. 2. NGS PANELS For NGS panels, several genes associated with the same phenotype are simultaneously sequenced. These panels provide increased diagnostic capability with a significantly reduced turnaround time and cost. CGC Genetics has several NGS panels for Ophthalmology that are constantly updated (www.cgcgenetics.com). Any gene studied in exome or NGS panel can also be individually sequenced and analyzed for deletion/duplication events. 3. EXPERTISE IN MEDICAL GENETICS CGC Genetics has Medical Geneticists specialized in genetic counseling for ophthalmological diseases who may advice in choosing the most appropriate -

WES Gene Package Multiple Congenital Anomalie.Xlsx
Whole Exome Sequencing Gene package Multiple congenital anomalie, version 5, 1‐2‐2018 Technical information DNA was enriched using Agilent SureSelect Clinical Research Exome V2 capture and paired‐end sequenced on the Illumina platform (outsourced). The aim is to obtain 8.1 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA‐MEM algorithm (reference: http://bio‐bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x A4GALT [Blood group, P1Pk system, P(2) phenotype], 111400 607922 101 100 100 99 [Blood group, P1Pk system, p phenotype], 111400 NOR polyagglutination syndrome, 111400 AAAS Achalasia‐addisonianism‐alacrimia syndrome, 231550 605378 73 100 100 100 AAGAB Keratoderma, palmoplantar, -

Psykisk Utviklingshemming Og Forsinket Utvikling
Psykisk utviklingshemming og forsinket utvikling Genpanel, versjon v03 Tabellen er sortert på gennavn (HGNC gensymbol) Navn på gen er iht. HGNC >x10 Andel av genet som har blitt lest med tilfredstillende kvalitet flere enn 10 ganger under sekvensering x10 er forventet dekning; faktisk dekning vil variere. Gen Gen (HGNC Transkript >10x Fenotype (symbol) ID) AAAS 13666 NM_015665.5 100% Achalasia-addisonianism-alacrimia syndrome OMIM AARS 20 NM_001605.2 100% Charcot-Marie-Tooth disease, axonal, type 2N OMIM Epileptic encephalopathy, early infantile, 29 OMIM AASS 17366 NM_005763.3 100% Hyperlysinemia OMIM Saccharopinuria OMIM ABCB11 42 NM_003742.2 100% Cholestasis, benign recurrent intrahepatic, 2 OMIM Cholestasis, progressive familial intrahepatic 2 OMIM ABCB7 48 NM_004299.5 100% Anemia, sideroblastic, with ataxia OMIM ABCC6 57 NM_001171.5 93% Arterial calcification, generalized, of infancy, 2 OMIM Pseudoxanthoma elasticum OMIM Pseudoxanthoma elasticum, forme fruste OMIM ABCC9 60 NM_005691.3 100% Hypertrichotic osteochondrodysplasia OMIM ABCD1 61 NM_000033.3 77% Adrenoleukodystrophy OMIM Adrenomyeloneuropathy, adult OMIM ABCD4 68 NM_005050.3 100% Methylmalonic aciduria and homocystinuria, cblJ type OMIM ABHD5 21396 NM_016006.4 100% Chanarin-Dorfman syndrome OMIM ACAD9 21497 NM_014049.4 99% Mitochondrial complex I deficiency due to ACAD9 deficiency OMIM ACADM 89 NM_000016.5 100% Acyl-CoA dehydrogenase, medium chain, deficiency of OMIM ACADS 90 NM_000017.3 100% Acyl-CoA dehydrogenase, short-chain, deficiency of OMIM ACADVL 92 NM_000018.3 100% VLCAD -

Use of Next Generation Sequencing for Isolated and Syndromic Anophthalmia, Microphthalmia and Coloboma (MAC): a New Approach to Molecular Genetic Diagnosis
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archivio della ricerca- Università di Roma La Sapienza Use of Next Generation Sequencing for isolated and syndromic Anophthalmia, Microphthalmia and Coloboma (MAC): a new approach to molecular genetic diagnosis Department of Cellular Biotechnologies and Hematology PhD in Human Biology and Medical Genetics XXX° cycle 2014-2017 Candidate Dr. Iapichino Giuseppe 1235269 Supervisor Correlator Prof. Pizzuti Antonio Prof. Novelli Antonio A/A 2016/2017 1. INTRODUCTION 1.1 Eye embryogenesis 1.2 MAC: Mixed group of diseases 1.2.1 Microphthalmia, Anophthalmia and Coloboma 1.2.2 Syndromic diseases: CHARGE syndrome Cornelia de Lange syndrome Axenfeld-Rieger syndrome Lenz’s Microphtalmia Papillorenal syndrome 1.3 Molecular Diagnosis: Next Generation Sequencing 2. AIM OF THE STUDY 3. MATERIALS AND METHODS 3.1 Patients Recruitment 3.2 Molecular Analysis 3.2.1 Blood collection, DNA extraction and quantification 3.2.2 NGS panel construction: Design Studio 3.2.3 NextSeq-500: features and workflow Libraries preparation: Nextera Rapid Capture Custom Enrichment Cluster generation Sequencing 3.2.4 Sanger Sequencing 3.2.5 Data Analysis 4. RESULTS 5. DISCUSSION 6. CONCLUSION 7. BIBLIOGRAPHY 1. INTRODUCTION 1.1 The eye: embryogenesis Eye development begins around the 4th week of gestation and can be summarized in four main stages [Zagozewski J.L. et al.; 2014]: Optic vesicle formation Induction of crystalline lens Retinal tissue formation Optic fissure closure Each of these events is highly regulated and coordinated by various transcription factors and circulating molecules [Adler R. et al.; 2007]. -

Absence of Mutations in PAX6 Gene in Three Cases of Morning Glory Syndrome Associated with Isolated Growth Hormone Defi Ciency
Absence of Mutations in PAX6 Gene in Three Cases of Morning Glory Syndrome Associated with Isolated Growth Hormone Defi ciency ABSTRACT clinical case report Morning glory syndrome (MGS) is a congenital optic disc dysplasia often as- sociated with craniofacial anomalies, especially basal encephalocele and hy- popituitarism. Clinical signs are varied and often occult. The PAX6 gene is involved in ocular morphogenesis and is expressed in numerous ocular tissues during development especially in the developing central nervous system. The GIL GUERRA-JUNIOR aim of the present study is to evaluate PAX6 in MGS associated with isolated ANGELA MARIA SPINOLA-CASTRO growth hormone defi ciency. Three pre-pubertal males (A, B and C) with MGS and short stature due to growth hormone defi ciency, treated with recombinant ADRIANA A. SIVIERO-MIACHON human growth hormone with limited response, were reported. Two of them ROBERTO GOMES NOGUEIRA had basal encephalocele. Coding and non-coding sequences corresponding of SOFIA HELENA V. L EMOS-MARINI PAX6 different transcripts were analyzed by direct sequencing. Nucleotide vari- ations causing putative aminoacid change were not observed. Patient A pre- LILIA FREIRE RODRIGUES D’SOUZA-LI sented the new IVS2+9G>A transition, whereas patients A and C were PRISCILA CRISTINA DA SILVA heterozygous for known single nucleotide polymorphisms (SNP) within the in- EMERSON SALVADOR S. FRANÇA tron 4. In addition, two SNP heterozygoses were observed for patient C in both intron 9 and 13. Sequencing also revealed several nucleotide variations in pa- FERNANDA CAROLINE SOARDI tient B. Two heterozygoses for known polymorphisms were identifi ed along MARICILDA PALANDI DE MELLO with a novel C>A nucleotide change in intron 4. -

Interactions of Pharmaceutical Companies with World Countries, Cancers and Rare Diseases from Wikipedia Network Analysis
RESEARCH ARTICLE Interactions of pharmaceutical companies with world countries, cancers and rare diseases from Wikipedia network analysis 1 1 2 3 Guillaume Rollin , Jose LagesID *, Tatiana S. SerebriyskayaID , Dima L. ShepelyanskyID 1 Institut UTINAM, CNRS, UMR 6213, OSU THETA, Universite de Bourgogne Franche-ComteÂ, BesancËon, France, 2 Laboratory for Translational Research and Personalized Medicine, Moscow Institute of Physics and Technology, Moscow, Russia, 3 Laboratoire de Physique TheÂorique, IRSAMC, Universite de Toulouse, CNRS, UPS, 31062 Toulouse, France a1111111111 a1111111111 * [email protected] a1111111111 a1111111111 a1111111111 Abstract Using the English Wikipedia network of more than 5 million articles we analyze interactions and interlinks between the 34 largest pharmaceutical companies, 195 world countries, 47 OPEN ACCESS rare renal diseases and 37 types of cancer. The recently developed algorithm using a Citation: Rollin G, Lages J, Serebriyskaya TS, reduced Google matrix (REGOMAX) allows us to take account both of direct Markov transi- Shepelyansky DL (2019) Interactions of tions between these articles and also of indirect transitions generated by the pathways pharmaceutical companies with world countries, between them via the global Wikipedia network. This approach therefore provides a com- cancers and rare diseases from Wikipedia network pact description of interactions between these articles that allows us to determine the analysis. PLoS ONE 14(12): e0225500. https://doi. org/10.1371/journal.pone.0225500 friendship networks between them, as well as the PageRank sensitivity of countries to phar- maceutical companies and rare renal diseases. We also show that the top pharmaceutical Editor: Aram Galstyan, USC Information Sciences Institute, UNITED STATES companies in terms of their Wikipedia PageRank are not those with the highest market capitalization. -

Targeted Exome Sequencing Identifies PBX1 As Involved
BASIC RESEARCH www.jasn.org Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract † ‡ | | Laurence Heidet,* Vincent Morinière,* Charline Henry,§ Lara De Tomasi,§ ¶ | | ‡†† Madeline Louise Reilly,§ ¶ Camille Humbert,§ Olivier Alibeu,** Cécile Fourrage, †† †† †† Christine Bole-Feysot,** Patrick Nitschké, Frédéric Tores, Marc Bras, |‡‡ || Marc Jeanpierre, Christine Pietrement,§§ Dominique Gaillard, Marie Gonzales,¶¶ ††† ‡‡‡ ||| Robert Novo,*** Elise Schaefer, Joëlle Roume, Jelena Martinovic,§§§ Valérie Malan, † | | ‡ | | Rémi Salomon,* § Sophie Saunier,§ Corinne Antignac, § and Cécile Jeanpierre§ *Assistance Publique - Hôpitaux de Paris, Centre de référence des Maladies Rénales Héréditaires de l’Enfant et de l’Adulte, Paris, France; †Assistance Publique - Hôpitaux de Paris, Service de Néphrologie Pédiatrique, ‡Assistance Publique - Hôpitaux de Paris, Département de Génétique, and |||Assistance Publique - Hôpitaux de Paris, Service de Cytogénétique, Hôpital Universitaire Necker- Enfants malades, Paris, France; §Institut National de la Santé et de la Recherche Médicale Unité Mixte de Recherche 1163, Laboratory of Hereditary Kidney Diseases, **Genomic Platform, Institut National de la Santé et de la Recherche Médicale Unité Mixte de Recherche 1163, Paris Descartes Sorbonne Paris Cité University, and ††Bioinformatic Plateform, Paris Descartes Sorbonne Paris Cité University, Imagine Institute, Paris, France; |Paris Descartes Sorbonne Paris Cité University, Paris, France; ¶Paris -

Nyresykdommer V01
2/1/2021 Nyresykdommer v01 Avdeling for medisinsk genetikk Nyresykdommer Genpanel, versjon v01 * Enkelte genomiske regioner har lav eller ingen sekvensdekning ved eksomsekvensering. Dette skyldes at de har stor likhet med andre områder i genomet, slik at spesifikk gjenkjennelse av disse områdene og påvisning av varianter i disse områdene, blir vanskelig og upålitelig. Disse genetiske regionene har vi identifisert ved å benytte USCS segmental duplication hvor områder større enn 1 kb og ≥90% likhet med andre regioner i genomet, gjenkjennes (https://genome.ucsc.edu). Vi gjør oppmerksom på at ved identifiseringav ekson oppstrøms for startkodon kan eksonnummereringen endres uten at transkript ID endres. Avdelingens websider har en full oversikt over områder som er affisert av segmentale duplikasjoner. ** Transkriptets kodende ekson. Ekson Gen Gen affisert (HGNC (HGNC Transkript Ekson** Fenotype av symbol) ID) segdup* ACE 2707 NM_000789.3 1-25 Renal tubular dysgenesis OMIM ACTG2 145 NM_001615.3 2-9 Visceral myopathy OMIM ACTN4 166 NM_004924.5 20-21 1-21 Glomerulosclerosis, focal segmental, 1 OMIM ADAMTS13 1366 NM_139025.4 1-29 Thrombotic thrombocytopenic purpura, familial OMIM ADCK4 19041 NM_024876.3 2-15 Nephrotic syndrome, type 9 OMIM AGT 333 NM_000029.3 2-5 Renal tubular dysgenesis OMIM AGTR1 336 NM_031850.3 3-4 Renal tubular dysgenesis OMIM file:///data/Nyre_v01-web.html 1/17 2/1/2021 Nyresykdommer v01 Ekson Gen Gen affisert (HGNC (HGNC Transkript Ekson** Fenotype av symbol) ID) segdup* AGXT 341 NM_000030.2 1-11 Hyperoxaluria, primary, type 1 OMIM -

Ocular Findings and Implications of Genetic Testing for Papillorenal Syndrome
Current Trends in Ophthalmology and Visual Science Hartnett M and Kaufman EJ. Curr Trends Ophthalmol and Vis Sci: CTOVS-101. Case Report DOI: 10.29011/CTOVS-101. 100001 Ocular Findings and Implications of Genetic Testing for Papillorenal Syndrome Magen Hartnett1, Evan J. Kaufman2* 1Department of Optometry Extern, New England College of Optometry, Virginia, USA 2Department of Ophthalmology, University of Virginia Health System, Virginia, USA *Corresponding author: Evan J. Kaufman, Department of Ophthalmology, University of Virginia Health System, Virginia, USA. Tel: +14349720070; Fax: +14349245180; Email: [email protected] Citation: Hartnett M, Kaufman EJ (2018) Ocular Findings and Implications of Genetic Testing for Papillorenal Syndrome. Curr Trends Ophthalmol and Vis Sci: CTOVS-101. DOI: 10.29011/CTOVS-101. 100001 Received Date: 17 March, 2018; Accepted Date: 04 May, 2018; Published Date: 11 May, 2018 Abstract Renal coloboma syndrome, also referred to as papillorenal syndrome, is a rare hereditary disorder that causes bilateral malformation of the optic nerve heads as well as a form of renal impairment [1,2]. It generally is acquired through autosomal dominant family inheritance, but cases of mutated forms have been reported in the absence of a family history. A 19-year-old female was seen at the University of Virginia’s Eye Clinic with bilateral optic disc anomalies and documented end stage renal disease (ESRD). She presented with a complaint of persistent blurring in both eyes that had not been treated previously. Clinical examination revealed refractive error, optic nerve dysplasia, and macular schisis of both eyes. This case outlines the clinical findings associated with apparent renal coloboma syndrome and its relation to genetic disorder. -
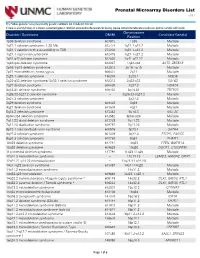
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome