Congenital Malformations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Blueprint Genetics Comprehensive Skeletal Dysplasias and Disorders
Comprehensive Skeletal Dysplasias and Disorders Panel Test code: MA3301 Is a 251 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of disorders involving the skeletal system. About Comprehensive Skeletal Dysplasias and Disorders This panel covers a broad spectrum of skeletal disorders including common and rare skeletal dysplasias (eg. achondroplasia, COL2A1 related dysplasias, diastrophic dysplasia, various types of spondylo-metaphyseal dysplasias), various ciliopathies with skeletal involvement (eg. short rib-polydactylies, asphyxiating thoracic dysplasia dysplasias and Ellis-van Creveld syndrome), various subtypes of osteogenesis imperfecta, campomelic dysplasia, slender bone dysplasias, dysplasias with multiple joint dislocations, chondrodysplasia punctata group of disorders, neonatal osteosclerotic dysplasias, osteopetrosis and related disorders, abnormal mineralization group of disorders (eg hypopohosphatasia), osteolysis group of disorders, disorders with disorganized development of skeletal components, overgrowth syndromes with skeletal involvement, craniosynostosis syndromes, dysostoses with predominant craniofacial involvement, dysostoses with predominant vertebral involvement, patellar dysostoses, brachydactylies, some disorders with limb hypoplasia-reduction defects, ectrodactyly with and without other manifestations, polydactyly-syndactyly-triphalangism group of disorders, and disorders with defects in joint formation and synostoses. Availability 4 weeks Gene Set Description -

With a Learning Disability; Guidance for General Practice
Improving identification of people with a learning disability: guidance for general practice NHS England and Improvement Publishing Approval Reference: 001030 Version 1 NHS England and NHS Improvement Contents Introduction .................................................................................... 2 Actions for practices ....................................................................... 4 Appendix 1: List of codes that indicate a learning disability ........... 7 Appendix 2: List of codes that may indicate a learning disability . 14 Appendix 3: List of outdated codes .............................................. 20 Appendix 4: Learning disability identification check-list ............... 22 1 | Contents Introduction 1. The NHS Long Term Plan1 commits to improve uptake of the existing annual health check in primary care for people aged over 14 years with a learning disability, so that at least 75% of those eligible have a learning disability health check each year. 2. There is also a need to increase the number of people receiving the annual seasonal flu vaccination, given the level of avoidable mortality associated with respiratory problems. 3. In 2017/18, only 44.6% of patients with a learning disability received a flu vaccination and only 55.1% of patients with a learning disability received an annual learning disability health check.2 4. In June 2019, NHS England and NHS Improvement announced a series of measures to improve coverage of annual health checks and flu vaccination for people with a learning disability. One of the commitments was to improve the quality of registers for people with a learning disability3. Clinical coding review 5. Most GP practices have developed a register of their patients known to have a learning disability. This has been developed from clinical diagnoses, from information gathered from learning disabilities teams and social services and has formed the basis of registers for people with learning disability developed for the Quality and Outcomes Framework (QOF). -

Craniosynostosis Precision Panel Overview Indications Clinical Utility
Craniosynostosis Precision Panel Overview Craniosynostosis is defined as the premature fusion of one or more cranial sutures, often resulting in abnormal head shape. It is a developmental craniofacial anomaly resulting from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). As well, craniosynostosis can be simple when only one suture fuses prematurely or complex/compound when there is a premature fusion of multiple sutures. Complex craniosynostosis are usually associated with other body deformities. The main morbidity risk is the elevated intracranial pressure and subsequent brain damage. When left untreated, craniosynostosis can cause serious complications such as developmental delay, facial abnormality, sensory, respiratory and neurological dysfunction, eye anomalies and psychosocial disturbances. In approximately 85% of the cases, this disease is isolated and nonsyndromic. Syndromic craniosynostosis usually present with multiorgan complications. The Igenomix Craniosynostosis Precision Panel can be used to make a directed and accurate diagnosis ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. Indications The Igenomix Craniosynostosis Precision Panel is indicated for those patients with a clinical diagnosis or suspicion with or without the following manifestations: ‐ Microcephaly ‐ Scaphocephaly (elongated head) ‐ Anterior plagiocephaly ‐ Brachycephaly ‐ Torticollis ‐ Frontal bossing Clinical Utility The clinical utility of this panel is: - The genetic and molecular confirmation for an accurate clinical diagnosis of a symptomatic patient. - Early initiation of treatment in the form surgical procedures to relieve fused sutures, midface advancement, limited phase of orthodontic treatment and combined 1 orthodontics/orthognathic surgery treatment. -

Primordial Dwarfism: a Case Series from North East of Iran and Literature Review
April 2019, Volume 7, Issue 2, Number 14 A Case Report and Literature Review: Primordial Dwarfism: A Case Series From North East of Iran and Literature Review Rahim Vakili1, 2 , Somayyeh Hashemian1* 1. Department of Pediatrics, Faculty of Medicine, Imam Reza Hospital, Mashhad University of Medical Sciences, Mashhad, Iran. 2. Medical Genetic Research Center, Mashhad University of Medical Sciences, Mashhad, Iran. Use your device to scan and read the article online Citation: Vakili R, Hashemian S. Primordial Dwarfism: A Case Series From North East of Iran and Literature Review. Journal of Pediatrics Review. 2019; 7(2):113-120. http://dx.doi.org/10.32598/jpr.7.2.113 : http://dx.doi.org/10.32598/jpr.7.2.113 A B S T R A C T Introduction: Primordial dwarfism is a rare class of genetic disorders, characterized by intrauterine See Page 119 Funding: growth retardation, short stature at birth and growth deficiency that persist throughout life. This disorder is caused by various mechanisms such as chromosomal abnormalities, molecular Article info: changes and mutation of genes that result in developmental defects, facial dysmorphism and Received: 15 February 2018 skeletal abnormalities in fetus. Primordial dwarfism includes 5 specific subtypes that their First Revision: 10 March 2018 descriptions vary from one type to another. This study aimed to report 7 cases of primordial Accepted: 30 May 2018 dwarfism as the first case series study and literature review of this disorder in Iran. Published: 01 April 2019 Case Presentations: This study presented primordial dwarfism patients and summarized clinical findings of 7 cases who referred to Pediatric endocrine wards in Imam Reza Hospital, from June 2016 to September 2017. -

Early ACCESS Diagnosed Conditions List
Iowa Early ACCESS Diagnosed Conditions Eligibility List List adapted with permission from Early Intervention Colorado To search for a specific word type "Ctrl F" to use the "Find" function. Is this diagnosis automatically eligible for Early Medical Diagnosis Name Other Names for the Diagnosis and Additional Diagnosis Information ACCESS? 6q terminal deletion syndrome Yes Achondrogenesis I Parenti-Fraccaro Yes Achondrogenesis II Langer-Saldino Yes Schinzel Acrocallosal syndrome; ACLS; ACS; Hallux duplication, postaxial polydactyly, and absence of the corpus Acrocallosal syndrome, Schinzel Type callosum Yes Acrodysplasia; Arkless-Graham syndrome; Maroteaux-Malamut syndrome; Nasal hypoplasia-peripheral dysostosis-intellectual disability syndrome; Peripheral dysostosis-nasal hypoplasia-intellectual disability (PNM) Acrodysostosis syndrome Yes ALD; AMN; X-ALD; Addison disease and cerebral sclerosis; Adrenomyeloneuropathy; Siemerling-creutzfeldt disease; Bronze schilder disease; Schilder disease; Melanodermic Leukodystrophy; sudanophilic leukodystrophy; Adrenoleukodystrophy Pelizaeus-Merzbacher disease Yes Agenesis of Corpus Callosum Absence of the corpus callosum; Hypogenesis of the corpus callosum; Dysplastic corpus callosum Yes Agenesis of Corpus Callosum and Chorioretinal Abnormality; Agenesis of Corpus Callosum With Chorioretinitis Abnormality; Agenesis of Corpus Callosum With Infantile Spasms And Ocular Anomalies; Chorioretinal Anomalies Aicardi syndrome with Agenesis Yes Alexander Disease Yes Allan Herndon syndrome Allan-Herndon-Dudley -

A Unifying Hypothesis on the Pathogenesis of Moyamoya Disease Based on a Systematic Review
NEUROSURGICAL FOCUS Neurosurg Focus 51 (3):E6, 2021 The mechanobiological theory: a unifying hypothesis on the pathogenesis of moyamoya disease based on a systematic review Bhanu Jayanand Sudhir, MCh,1 Arun Gowda Keelara, MS,1 Easwer Harihara Venkat, MCh,1 Ken Kazumata, MD, PhD,2 and Ananthalakshmy Sundararaman, PhD3 1Department of Neurosurgery, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Trivandrum, Kerala State, India; 2Department of Neurosurgery, Hokkaido University Graduate School of Medicine, Sapporo, Japan; and 3Department of Cardiovascular Diseases and Diabetes Biology, Rajiv Gandhi Centre for Biotechnology, Trivandrum, Kerala State, India OBJECTIVE Moyamoya angiopathy (MMA) affects the distal internal carotid artery and is designated as moyamoya disease (MMD) when predisposing conditions are absent, or moyamoya syndrome (MMS) when it occurs secondary to other causes. The authors aimed to investigate the reason for this anatomical site predilection of MMA. There is compel- ling evidence to suggest that MMA is a phenomenon that occurs due to stereotyped mechanobiological processes. Literature regarding MMD and MMS was systematically reviewed to decipher a common pattern relating to the develop- ment of MMA. METHODS A systematic review was conducted to understand the pathogenesis of MMA in accordance with PRISMA guidelines. PubMed MEDLINE and Scopus were searched using “moyamoya” and “pathogenesis” as common keywords and specific keywords related to six identified key factors. Additionally, a literature search was performed for MMS using “moyamoya” and “pathogenesis” combined with reported associations. A progressive search of the literature was also performed using the keywords “matrix metalloprotease,” “tissue inhibitor of matrix metalloprotease,” “endothelial cell,” “smooth muscle cell,” “cytokines,” “endothelin,” and “transforming growth factor” to infer the missing links in molecular pathogenesis of MMA. -

Cilia in Hereditary Cerebral Anomalies Sophie Thomas, Lucile Boutaud, Madeline Louise Reilly, Alexandre Benmerah
Cilia in hereditary cerebral anomalies Sophie Thomas, Lucile Boutaud, Madeline Louise Reilly, Alexandre Benmerah To cite this version: Sophie Thomas, Lucile Boutaud, Madeline Louise Reilly, Alexandre Benmerah. Cilia in hereditary cerebral anomalies. Biology of the Cell, Wiley, 2019, 10.1111/boc.201900012. inserm-02263786 HAL Id: inserm-02263786 https://www.hal.inserm.fr/inserm-02263786 Submitted on 8 Aug 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Cilia in hereditary cerebral anomalies Sophie Thomas1,*, Lucile Boutaud1, Madeline Louise Reilly2,3, and Alexandre Benmerah2,* 1Laboratory of Embryology and Genetics of Human Malformation, INSERM UMR 1163, Paris Descartes University, Imagine Institute, 75015 Paris, France. 2Laboratory of Hereditary Kidney Diseases, INSERM UMR 1163, Paris Descartes University, Imagine Institute, 75015 Paris, France. 3Paris Diderot University, 75013 Paris, France. * To whom correspondence should be addressed: Alexandre Benmerah, Institut Imagine, 24 boulevard du Montparnasse, 75015 PARIS, France. Tel: +33 1 42 75 43 44, fax: +33 1 42 75 42 25, email: [email protected] Sophie Thomas, Institut Imagine, 24 boulevard du Montparnasse, 75015 PARIS, France. Tel : +33 +33 1 42 75 43 10, fax: +33 1 42 75 42 25, email: [email protected] 1 Abstract: Ciliopathies are complex genetic multisystem disorders causally related to abnormal assembly or function of motile or non-motile cilia. -

To Be Your Local Expert – a General Pediatrician’S Story
To Be Your Local Expert – A General Pediatrician’s Story DDC Clinic mission: To enhance the quality of life for people with special needs caused by rare genetic disorders What Does It Take to Be Your Local Expert? “Through My Window …” - “Research Clinic Opens in Ohio for Genetic Maladies That Haunt Amish Families” By FRANCIS X. CLINES June 20, 2002 “You Have a Lot to Learn!” Chicken breast disease – Amish Normaline myopathy • Autosomal recessive • Onset with neonatal tremors and hypotonia • Contractures • Chest deformity • Respiratory failure To be your local expert is not simply a desire or a choice, it is a call of duty! Cohen Syndrome – Overview • Genetics: autosomal recessive • First reported by Dr. Michael Cohen in 1968 • Gene related to the disease: COH1 (VPS13B) on chromosome 8q22 • Incidence: Over 1000 patients are diagnosed worldwide, and over 200 patients are reported • A disease with multiple system involvements 7th Cohen Syndrome Family Gathering in June 2014 Cohen Syndrome – Clinical Manifestations • Psychomotor retardation • Microcephaly • Characteristic facial features • Childhood hypotonia and joint laxity • Progressive retinochoroidal dystrophy and myopia • Truncal obesity • Neutropenia • Cheerful disposition • Any earlier signs or symptoms? Cohen Syndrome – Fetal Movements in 65 Patients Cohen Syndrome – Facial Features • Thick hair and low hairline • High -arched and waved-shaped eyelids • Long and thick eyelashes, thick eyebrows • High and narrow palate • Short philtrum and prominent upper central incisors, resulting -

A Systematic Review and Qualitative Synthesis
This is a repository copy of The experiences of fathers of children with a life-limiting condition: a systematic review and qualitative synthesis. White Rose Research Online URL for this paper: https://eprints.whiterose.ac.uk/174916/ Version: Accepted Version Article: Fisher, Victoria orcid.org/0000-0002-1497-1241, Fraser, Lorna Katharine orcid.org/0000- 0002-1360-4191 and Taylor, Jo orcid.org/0000-0001-5898-0900 (Accepted: 2021) The experiences of fathers of children with a life-limiting condition: a systematic review and qualitative synthesis. BMJ Supportive & Palliative Care. ISSN 2045-4368 (In Press) Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Supplemental material; search strategy for Medline 1. Fathers/ 2. father*.tw. 3. dad*.tw. 4. stepfather*.tw. 5. step-father*.tw. 6. (foster* adj3 father*).tw. 7. (adopt* adj3 father*).tw. 8. 1 or 2 or 3 or 4 or 5 or 6 or 7 9. -
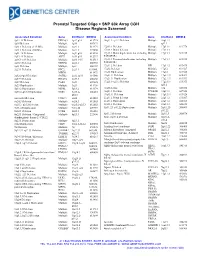
180K ISCA Array
Prenatal Targeted Oligo + SNP 60k Array CGH Disease Regions Screened Associated Condition Gene Chr/Band OMIM # Associated Condition Gene Chr/Band OMIM # 1p32-31 Deletion DIRAS3 1p32-p31 613735 16q11.2–q12.2 Deletion Multiple 16q11.2– 1p36 Deletion Multiple 1p36 607872 q12.2 1q21.1 Deletion (1.35 Mb) Multiple 1q21.1 612474 17p13.1 Deletion Multiple 17p13.1 613776 1q21.1 Deletion (200 Kb) Multiple 1q21.1 274000 17p13.3 Distal deletion Multiple 17p13.3 1q41-42 Deletion Multiple 1q41-q42 612530 17p13.3 Distal duplication, not including Multiple 17p13.3 613215 1q43-44 Deletion AKT3 1q43-q44 612337 PAFAH1B1 2p16.1-p15 Deletion Multiple 2p16.1-15 612513 17p13.3 Proximal duplication, including Multiple 17p13.3 613215 2p16.3 Deletion NRXN1 2p16.3 600565 PAFAH1B1 2p21 Deletion Multiple 2p21 606407 17q11.2 Deletion NF1 17q11.2 613675 2q23.1 Deletion EPC2, 2q23.1 611000, 17q12 Deletion Multiple 17q12 614527 MBD5 611472 17q12 Duplication Multiple 17q12 614526 2q32.2–q33 Deletion SATB2 2q32.2q33 119540 17q21.31 Deletion Multiple 17q21.31 610443 2q37.3 Deletion HDAC4 2q37.3 600430 17q21.31 Duplication Multiple 17q21.31 613533 3q29 Deletion PAK2 3q29 609425 17q23.1–q23.2 Deletion Multiple 17q23.1- 613355 3q29 Duplication Multiple 3q29 611936 q23.2 5p13.2 Duplication NIPBL 5p13.2 613174 18q Deletion Multiple 18q 601808 5q35.2–q35.3 Duplication NSD1 5q35.2– 606681 18q21.1 Deletion TCEB3B 18q21.1 609522 q35.3 19q13.11 Deletion Multiple 19q13.11 613026 6pter-p24 Deletion FOXC1 6p24 612582 22q11.2 Distal deletion Multiple 22q11.2 611867 6q24.3 Deletion Multiple -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G. Phd, Mphil, Dclinpsychol) at the University of Edinburgh
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Expanding the genetics of microcephalic primordial dwarfism Jennie Elaine Murray Thesis submitted for the degree of Doctor of Philosophy The University of Edinburgh October 2014 This thesis is composed of original research undertaken by myself, and where the work of others is included their contributions have been duly acknowledged. This work has not been submitted for any other degree or professional qualification. Jennie Murray 2 Acknowledgements Firstly, I would like to say a big thank you to my supervisor, Professor Andrew Jackson, for his fantastic guidance and support over the last 3 years. Your integrity and dedication has been inspiring and, although I still have a long way to go, your input has been invaluable in helping me grow and develop as a clinical scientist.