Vicinal Oxidative Electrochemical Alkene Difunctionalization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Arenechromium Tricarbonyl Complexes: Conformational
η6 – ARENECHROMIUM TRICARBONYL COMPLEXES: CONFORMATIONAL ANALYSIS, STEREOCONTROL IN NUCLEOPHILIC ADDITION AND APPLICATIONS IN ORGANIC SYNTHESIS by HARINANDINI PARAMAHAMSAN Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Thesis Advisor: Prof. Anthony J. Pearson Department of Chemistry CASE WESTERN RESERVE UNIVERSITY May 2005 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the dissertation of Harinandini Paramahamsan candidate for the Ph.D. degree*. (signed) Prof. Philip P. Garner (Chair of the Committee, Department of Chemistry, CWRU) Prof. Anthony J. Pearson (Department of Chemistry, CWRU) Prof. Fred L. Urbach (Department of Chemistry, CWRU) Dr. Zwong-Wu Guo (Department of Chemistry, CWRU) Dr. Stuart J. Rowan (Department of Macromolecular Science and Engineering, CWRU) Date: 14th January 2005 *We also certify that written approval has been obtained for any propriety material contained therein. To Amma, Naina & all my Teachers Table of Contents List of Tables………………………………………………………………………..……iv List of Figures…………………………………………………………………….…........vi List of Schemes…………………………………………………………………….….….ix List of Equations………………………………………………………...……….……….xi Acknowledgements………………………………………………………….…..……….xii List of Abbreviations……………………………………………………………………xiv Abstract………………………………………………………………………………….xvi CHAPTER I........................................................................................................................ 1 I.1 Structure and Bonding ........................................................................................... -

Transcription 11.12.07
Lab 17A • 12/07/11 [lab quiz] Nomenclature of alkenes The first molecule that I want to look at is this one, where we have the two methyl groups on one side, two hydrogens on the other side. Would it be appropriate to use cis or trans, or E or Z, or both, or neither? One carbon of the double bond versus the other, those are the two different sides of the double, then the top versus the bottom are the two faces of the double bond. If we notice, on both the top face and the bottom face, we have a methyl group that is pointed the same way as a hydrogen. There is a steric factor as far as what alkene would prefer to form thermodynamically, so there is an importance that there’s some interaction there. That methyl group with one hydrogen is exactly the same interaction as you’d have the methyl group and the other hydrogen pointed the opposite way – meaning that if you were to switch the two hydrogens, you’d end up with exactly the same molecule again. The only reason that we use the term cis or trans or E or Z is to describe that it is one configuration versus another, but since there’s only one configuration possible, there’s therefore no term that should be used. It would, in fact, be wrong to call this cis, trans, E, or Z. When an alkene has two of the same substituent on the same side, there is only one unique configuration of that alkene, and so it cannot be called cis, trans, E, or Z. -

Vicinal Difunctionalization of Carbonâ
ARTICLE https://doi.org/10.1038/s41467-020-19748-z OPEN Vicinal difunctionalization of carbon–carbon double bond for the platform synthesis of trifluoroalkyl amines ✉ Ferenc Béke1, Ádám Mészáros1, Ágnes Tóth1, Bence Béla Botlik1 & Zoltán Novák 1 Regioselective vicinal diamination of carbon–carbon double bonds with two different amines is a synthetic challenge under transition metal-free conditions, especially for the synthesis of 1234567890():,; trifluoromethylated amines. However, the synthesis of ethylene diamines and fluorinated amine compounds is demanded, especially in the pharmaceutical sector. Herein, we demonstrate that the controllable double nucleophilic functionalization of an activated alkene synthon, originated from a trifluoropropenyliodonium salt with two distinct nucleophiles, enables the selective synthesis of trifluoromethylated ethylene amines and diamines on broad scale with high efficiency under mild reaction conditions. Considering the chemical nature of the reactants, our synthetic approach brings forth an efficient methodology and provides versatile access to highly fluorinated amines. 1 ELTE “Lendület” Catalysis and Organic Synthesis Research Group, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter stny. 1/A, 1117 ✉ Budapest, Hungary. email: [email protected] NATURE COMMUNICATIONS | (2020) 11:5924 | https://doi.org/10.1038/s41467-020-19748-z | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-19748-z he vicinal diamine backbone is a prevalent motif in natural free conditions has been presented with bifunctional nucleophiles products, chelating agents, chiral ligands, and pharma- in an intramolecular manner to afford N,N- and N,O-heterocyclic T 1,2 ceuticals . Nature presents this structural motif in the systems using alkenyl-sulfonium salts with two adjacent electro- form of non-proteinogenic amino acids which constitute the philic centers29–32. -

Studies in Stereochemistry Donald Charles Best Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1968 Studies in stereochemistry Donald Charles Best Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Best, Donald Charles, "Studies in stereochemistry " (1968). Retrospective Theses and Dissertations. 3648. https://lib.dr.iastate.edu/rtd/3648 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microiilined exactly as received g 8"14.773 BEST, Donald Charles, 1940- STUDIES IN STEREOCHEMISTRY. Iowa State University, Ph,D., 1968 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan STUDIES IN STEREOCHEMISTRY by Donald Charles Best A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject : Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University of Science and Technology Ames, Iowa 1968 il TABLE OF CONTENTS Page PART I: CONFORMATIONAL PREFERENCES OF ACYCLIC PIASTEREOMERS la HISTORICAL lb I RESULTS AND DISCUSSION 7 EXPERIMENTAL 48 PART II : SUBSTITUENT EFFECTS ON SUSPECTED PHENONIUM ION INTERMEDIATES 82; HISTORICAL R3 RESULTS AND DISCUSSION 94 ' EXPERIMENTAL 109 LITERATURE CITED 155 ACKNOWLEDGEMENTS l40 ' la PART I: CONFORMATIONAL PREFERENCES OF ACYCLIC DIASTEREOMERS lb HISTORICAL The subject of conformational analysis was introduced about seventy-five years ago by Sachse (49). -

Alkenes and Alkynes I: Properties and Synthesis
Chapter 7 Alkenes and Alkynes I: Properties and Synthesis. Elimination Reactions of Alkyl Halides Ch. 7 - 1 1. Introduction Alkenes ● Hydrocarbons containing C=C ● Old name: olefins CH2OH Vitamin A H3C H3C H H Cholesterol HO Ch. 7 - 2 Alkynes ● Hydrocarbons containing C≡C ● Common name: acetylenes H N O I Cl C O C O Cl F3C C Cl C Cl Efavirenz Haloprogin (antiviral, AIDS therapeutic) (antifungal, antiseptic) Ch. 7 - 3 2. The (E) - (Z) System for Designating Alkene Diastereomers Cis-Trans System ● Useful for 1,2-disubstituted alkenes ● Examples: H Br Cl Cl (1) Br vs H H H trans -1-Bromo- cis -1-Bromo- 2-chloroethene 2-chloroethene Ch. 7 - 4 ● Examples H (2) vs H H H trans -3-Hexene cis -3-Hexene Br (3) Br Br vs Br trans -1,3- cis -1,3- Dibromopropene Dibromopropene Ch. 7 - 5 (E) - (Z) System ● Difficulties encountered for trisubstituted and tetrasubstituted alkenes CH3 e.g. Cl cis or trans? Br H Cl is cis to CH3 and trans to Br Ch. 7 - 6 The Cahn-Ingold-Prelog (E) - (Z) Convention ● The system is based on the atomic number of the attached atom ● The higher the atomic number, the higher the priority Ch. 7 - 7 The Cahn-Ingold-Prelog (E) - (Z) Convention ● (E) configuration – the highest priority groups are on the opposite side of the double bond “E ” stands for “entgegen”; it means “opposite” in German ● (Z) configuration – the highest priority groups are on the same side of the double bond “Z ” stands for “zusammer”; it means “together” in German Ch. -

Chem Soc Rev Tutorial Review
Please do not adjust margins Chem Soc Rev TUTORIAL REVIEW Stereoselective Arene Formation Achim Link and Christof Sparr* While aromatic hydrocarbons are ubiquitous in organic chemistry, they are typically not associated with chirality and stereoisomerism. Due to the planarity and symmetry of simple arenes, methods to assemble aromatic rings are therefore not routinely considered for the stereoselective synthesis of chiral compounds. The aim of this tutorial review is to contrast Received 15th December 2017, this common perception with the counterintuitive circumstance that stereoselective arene formation offers a means to Accepted stereoselectively prepare an exceptional range of chiral aromatic structures. The versatility of these methods across various types of molecular scaffolds allows to control stereocentre configuration, helical chiral compounds, the configuration of DOI: 10.1039/x0xx00000x rotationally restricted stereogenic axes, planar chiral molecules or curved polyaromatic systems. Furthermore, www.rsc.org/chemsocrev stereoselective arene formation holds great promise for the selective construction of extended- but structurally well-defined chiral structures. Key learning points: 1) Counterintuitively, arene formation offers a means to stereoselectively prepare a broad range of chiral structures. 2) Substrate desymmetrization by the de novo construction of an aromatic ring allows controlling stereocentre configuration 3) The non-planar shape of helicenes or curved aromatic systems can be controlled during arene assembly. 4) Rotationally restricted atropisomers and planar chiral compounds are readily accessible by stereoselective arene formation. forming reactions offer unique opportunities to prepare various 1. Introduction stereochemically complex compounds in isomerically enriched form, a concept that has recently gained momentum and Owing to the high symmetry of arene-archetypes such as emerged as an effective strategy also for the synthesis of benzene, naphthalene or anthracene, aromatic hydrocarbons extended structures. -

Infrared Tables (Short Summary of Common Absorption Frequencies)
Beauchamp Spectroscopy Tables 1 Infrared Tables (short summary of common absorption frequencies) The values given in the tables that follow are typical values. Specific bands may fall over a range of wavenumbers, cm-1. Specific substituents may cause variations in absorption frequencies. Absorption intensities may be stronger or weaker than expected, often depending on dipole moments. Additional bands may confuse the interpretation. In very symmetrical compounds there may be fewer than the expected number of absorption bands (it is even possible that all bands of a functional group may disappear, i.e. a symmetrically substituted alkyne!). Infrared spectra are generally informative about what functional groups are present, but not always. The 1H and 13C NMR’s are often just as informative about functional groups, and sometimes even more so in this regard. Information obtained from one spectroscopic technique should be verified or expanded by consulting the other spectroscopic techniques. IR Summary - All numerical values in the tables below are given in wavenumbers, cm-1 Bonds to Carbon (stretching wave numbers) sp3 C-X single bonds sp2 C-X single bonds CN CO CO CC CC CN 1000-1350 1050-1150 1100-1350 not used not very useful alkoxy C-O not very useful 1250 acyl and phenyl C-O sp2 C-X double bonds sp C-X triple bonds CO CC CN CC CN 1640-1810 expanded table 1600-1680 1640-1690 on next page 2100-2250 2240-2260 Stronger dipoles produce more intense IR bands and weaker dipoles produce less intense IR bands (sometimes none). Bonds to Hydrogen -

Stereochemistry
Lecture note- 2 Organic Chemistry CHE 502 STEREOCHEMISTRY DEPARTMENT OF CHEMISTRY UTTARAKHAND OPEN UNIVERSITY UNIT 4: STEREOCHEMISTRY 1 CONTENTS 4.1 Objectives 4.2 Introduction 4.3 Isomerism 4.4 Structural (Constitutional) Isomerism 4.5 Stereo (Configurational) isomerism 4.5.1 Geometrical Isomerism 4.5.2 Optical Isomerism 4.6 Element of Symmetry 4.7 Stereogenic centre (Stereogenicity) 4.7.1 Optical activity and Enantiomerism 4.7.2 Properties of enantiomerism 4.7.3 Chiral and achiral molecules with two stereogenic centers 4.7.4 Diastereomers 4.7.5 Properties of Diastereomers 4.7.6 Erythro (syn) Threo (anti) diastereomers 4.7.7 Meso compounds 4.8 Relative and absolute configurations 4.8.1 D/L nomenclature 4.8.2 R/S nomenclature 4.8.3 Sequence Rule 4.9 Newman and sawhorse projection formulae 4.10 Fisher flying and wedge formulae 4.11 Racemic mixture (racemates) 4.12 Quasi enantiomers 4.13 Quasi racemates 4.14 Stereochemistry of allenes, spiranes, biphenyls, ansa compounds, cyclophanes and related compounds 4.15 Summary 4.16 Terminal Questions 4.17 Answers 4.1 OBJECTIVES In this unit learner will be able to: ➢ Depict various types of isomerism exhibited by organic compounds and their representation ➢ Analyze the three dimensional depictions of organic compounds and their two dimensional representations. ➢ Learn Stereogenicity, chirality, enantiomerism, diastereomerism, their relative and absolute configurations ➢ Learn about the various stereo chemical descriptors such as (cis-trans, E/Z, D/L, d/l, erythro/threo, R/S and syn/anti) given to organic molecules differ ➢ Describe the stereochemistry of various rigid and complex molecules like spiranes, adamentanes, catenanes, cyclophanes etc. -

Applications of Radical Reactions for the Synthesis of -Amino Acids and Carbonyl Compounds
Applications of radical reactions for the synthesis of β-amino acids and carbonyl compounds Xuan Chen To cite this version: Xuan Chen. Applications of radical reactions for the synthesis of β-amino acids and carbonyl com- pounds. Organic chemistry. Institut Polytechnique de Paris, 2020. English. NNT : 2020IPPAX042. tel-02983194 HAL Id: tel-02983194 https://tel.archives-ouvertes.fr/tel-02983194 Submitted on 29 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Applications of radical reactions for the synthesis of β-amino acids and carbonyl compounds 42 0 Thèse de doctorat de l’Institut Polytechnique de Paris AX préparée à École Polytechnique IPP 20 20 École doctorale n°626 Ecole Doctorale de l’Institut Polytechnique de : Paris (ED IP Paris) Spécialité de doctorat: Chimie NNT Thèse présentée et soutenue à Palaiseau, le 15 Septembre 2020, par M. Xuan CHEN Composition du Jury : Isabelle GILLAIZEAU Professeur, Université d'Orléans (UMR 7311) Présidente Luc NEUVILLE Docteur, University Paris-Saclay Rapporteur Benoît CROUSSE Docteur, University Paris-Sud (UMR 8076) Rapporteur Samir Z. ZARD Professeur, Ecole Polytechnique (UMR 7652) Examinateur, Directeur de thèse Acknowledgements I First of all, I am going to express my gratitude to my supervisor Professor Samir Z. -

Hydrogenation of Alkenes – Addition of H-H (H2) to the Π-Bond of Alkenes to Afford an Alkane
Chapter 6: Reactions of Alkenes: Addition Reactions 6.1: Hydrogenation of Alkenes – addition of H-H (H2) to the π-bond of alkenes to afford an alkane. The reaction must be catalyzed by metals such as Pd, Pt, Rh, and Ni. H H H H Pd/C ΔH° = -136 KJ/mol + C C hydrogenation C C H H H EtOH H H H H H C-C π-bond H-H C-H = 243 KJ/mol = 435 KJ/mol = 2 x -410 KJ/mol = -142 KJ/mol • The catalysts is not soluble in the reaction media, thus this process is referred to as a heterogenous catalysis. • The catalyst assists in breaking the π-bond of the alkene and the H-H σ-bond. • The reaction takes places on the surface of the catalyst. Thus, the rate of the reaction is proportional to the surface area of the catalyst. 127 • Carbon-carbon π-bond of alkenes and alkynes can be reduced to the corresponding saturated C-C bond. Other π-bond bond such as C=O (carbonyl) and C≡N are not easily reduced by catalytic hydrogenation. The C=C bonds of aryl rings are not easily reduced. O O H2, PtO2 ethanol O H2, Pd/C C5H11 OH CH3(CH2)16CO2H Linoleic Acid (unsaturated fatty acid) Steric Acid (saturated fatty acid) O O H , Pd/C OCH3 2 OCH3 ethanol H , Pd/C C 2 C N N ethanol 128 6.2: Heats of Hydrogenation -an be used to measure relative stability of isomeric alkenes H H H CH3 trans isomer is ~3 KJ/mol H3C CH3 H3C H more stable than the cis-2-butene trans-2-butene cis isomer ΔH°combustion : -2710 KJ/mol -2707 KJ/mol H H H CH3 H2, Pd H2, Pd CH3CH2CH2CH3 H3C CH3 H3C H cis-2-butene trans-2-butene ΔH°hydrogenation: -119 KJ/mol -115 KJ/mol trans isomer is ~4 KJ/mol more stable than the cis isomer The greater release of heat, the less stable the reactant. -

You Must Have Your Answers Written in PERMANENT Ink If You Want a Regrade!!!! This Means No Test Written in Pencil Or ERASABLE INK Will Be Regraded
NAME (Print): _____________________________ Chemistry 310M/318M Dr. Brent Iverson 3rd Midterm SIGNATURE: _____________________________ November 17, 2011 Please print the first three letters of your last name in the three boxes Please Note: This test may be a bit long, but there is a reason. I would like to give you a lot of little questions, so you can find ones you can answer and show me what you know, rather than just a few questions that may be testing the one thing you forgot. I recommend you look the exam over and answer the questions you are sure of first, then go back and try to figure out the rest. Also make sure to look at the point totals on the questions as a guide to help budget your time. You must have your answers written in PERMANENT ink if you want a regrade!!!! This means no test written in pencil or ERASABLE INK will be regraded. Please note: We routinely xerox a number of exams following initial grading to guard against receiving altered answers during the regrading process. FINALLY, DUE TO SOME UNFORTUNATE RECENT INCIDENCTS YOU ARE NOT ALLOWED TO INTERACT WITH YOUR CELL PHONE IN ANY WAY. IF YOU TOUCH YOUR CELL PHONE DURING THE EXAM YOU WILL GET A "0" NO MATTER WHAT YOU ARE DOING WITH THE PHONE. PUT IT AWAY AND LEAVE IT THERE!!! Honor Code The core values of the University of Texas at Austin are learning, discovery, freedom, leadership, individual opportunity, and responsibility. Each member of the University is expected to uphold these values through integrity, honesty, trust, fairness, and respect toward peers and community. -
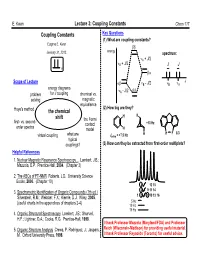
The Coupling Constant JHH Depends on the H-C-C-H Dihedral Halogens Are Both Electron-Withdrawing by Induction and Angle
E. Kwan Lecture 3: Coupling Constants Chem 117 Coupling Constants Key Questions (1) What are coupling constants? Eugene E. Kwan January 31, 2012. energy spectrum: A + J/2 B + J/2 J J Scope of Lecture B - J/2 B A energy diagrams - J/2 problem for J coupling chemical vs. A solving magnetic equivalence (2) How big are they? Hoye's method the chemical H H shift the Fermi first- vs. second- contact +10 Hz order spectra model H H H F 8.3 virtual coupling what are Jortho = +7.5 Hz typical couplings? (3) How can they be extracted from first-order multiplets? Helpful References 1. Nuclear Magnetic Resonance Spectroscopy... Lambert, J.B.; Mazzola, E.P. Prentice-Hall, 2004. (Chapter 3) 2. The ABCs of FT-NMR Roberts, J.D. University Science Books, 2000. (Chapter 10) 4 7 12 15 3. Spectrometric Identification of Organic Compounds (7th ed.) 3 6 9 11 14 Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Wiley, 2005. 125 8 10 13 16 5 Hz (useful charts in the appendices of chapters 2-4) 10 Hz 15 Hz 4. Organic Structural Spectroscopy Lambert, J.B.; Shurvell, H.F.; Lightner, D.A.; Cooks, R.G. Prentice-Hall, 1998. I thank Professor Mazzola (Maryland/FDA) and Professor 5. Organic Structure Analysis Crews, P. Rodriguez, J.; Jaspars, Reich (Wisconsin-Madison) for providing useful material. M. Oxford University Press, 1998. I thank Professor Reynolds (Toronto) for useful advice. E. Kwan Lecture 3: Coupling Constants Chem 117 Review: What's a Coupling Constant? These are the carbon-13 satellites.