Infrared Tables (Short Summary of Common Absorption Frequencies)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Origin of the Peculiarities of the Vietnamese Alphabet André-Georges Haudricourt
The origin of the peculiarities of the Vietnamese alphabet André-Georges Haudricourt To cite this version: André-Georges Haudricourt. The origin of the peculiarities of the Vietnamese alphabet. Mon-Khmer Studies, 2010, 39, pp.89-104. halshs-00918824v2 HAL Id: halshs-00918824 https://halshs.archives-ouvertes.fr/halshs-00918824v2 Submitted on 17 Dec 2013 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Published in Mon-Khmer Studies 39. 89–104 (2010). The origin of the peculiarities of the Vietnamese alphabet by André-Georges Haudricourt Translated by Alexis Michaud, LACITO-CNRS, France Originally published as: L’origine des particularités de l’alphabet vietnamien, Dân Việt Nam 3:61-68, 1949. Translator’s foreword André-Georges Haudricourt’s contribution to Southeast Asian studies is internationally acknowledged, witness the Haudricourt Festschrift (Suriya, Thomas and Suwilai 1985). However, many of Haudricourt’s works are not yet available to the English-reading public. A volume of the most important papers by André-Georges Haudricourt, translated by an international team of specialists, is currently in preparation. Its aim is to share with the English- speaking academic community Haudricourt’s seminal publications, many of which address issues in Southeast Asian languages, linguistics and social anthropology. -
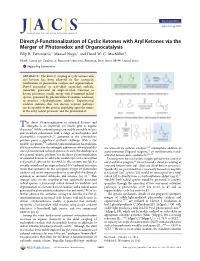
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

The Reaction of Aminonitriles with Aminothiols: a Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean
life Article The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean Ibrahim Shalayel , Seydou Coulibaly, Kieu Dung Ly, Anne Milet and Yannick Vallée * Univ. Grenoble Alpes, CNRS, Département de Chimie Moléculaire, Campus, F-38058 Grenoble, France; [email protected] (I.S.); [email protected] (S.C.); [email protected] (K.D.L.); [email protected] (A.M.) * Correspondence: [email protected] Received: 28 September 2018; Accepted: 18 October 2018; Published: 19 October 2018 Abstract: The Strecker reaction of aldehydes with ammonia and hydrogen cyanide first leads to α-aminonitriles, which are then hydrolyzed to α-amino acids. However, before reacting with water, these aminonitriles can be trapped by aminothiols, such as cysteine or homocysteine, to give 5- or 6-membered ring heterocycles, which in turn are hydrolyzed to dipeptides. We propose that this two-step process enabled the formation of thiol-containing dipeptides in the primitive ocean. These small peptides are able to promote the formation of other peptide bonds and of heterocyclic molecules. Theoretical calculations support our experimental results. They predict that α-aminonitriles should be more reactive than other nitriles, and that imidazoles should be formed from transiently formed amidinonitriles. Overall, this set of reactions delineates a possible early stage of the development of organic chemistry, hence of life, on Earth dominated by nitriles and thiol-rich peptides (TRP). Keywords: origin of life; prebiotic chemistry; thiol-rich peptides; cysteine; aminonitriles; imidazoles 1. Introduction In ribosomes, peptide bonds are formed by the reaction of the amine group of an amino acid with an ester function. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Aldehydes and Ketones Are Simple Organic Compounds Containing a Carbonyl Group
Aldehydes and Ketones are simple organic compounds containing a carbonyl group. Carbonyl group contains carbon- oxygen double bond. These organic compounds are simple because the carbon atom presents in the carbonyl group lack reactive groups such as OH or Cl. By Dr. Sayed Hasan Mehdi Assistant Professor Department of Chemistry Shia P.G. College, Lucknow Dr. S.Hasan Mehdi 6/13/2020 This is to bring to kind notice that the matter of this e- content is for the B.Sc. IV semester students. It has been taken from the following sources. The students are advised to follow these books as well. •A TEXTBOOK OF ORGANIC CHEMISTRY by Arun Bahl & B.S. Bahl, S. Chand & Company Ltd. Publication. •Graduate Organic Chemistry by M. K. Jain and S.C. Sharma, Vishal Publishing Co. •Pradeep’s Organic Chemistry Vol II by R. N. Dhawan, Pradeep Publication, Jalandhar. Dr. S.Hasan Mehdi 6/13/2020 An aldehyde is one of the classes of carbonyl group- containing alkyl group on one end and hydrogen on the other end. The R and Ar denote alkyl or aryl member respectively. In the condensed form, the aldehyde is written as –CHO. Dr. S.Hasan Mehdi 6/13/2020 Dr. S.Hasan Mehdi 6/13/2020 1. From Alcohols: a. By oxidation of Alcohols: Aldehydes and ketones can be prepared by the controlled oxidation of primary and secondary alcohols using an acidified solution of potassium dichromate or permanganate. Primary alcohol produces aldehydesRef. Last slide. O K Cr O RCH2OH + [O] 2 2 7 + R C H H 10 Alcohol Aldehyde O CH3CH2OH+ [O] K2Cr2O7 + CH3 C H H Ethyl Alcohol Acetaldehyde The aldehydes formed in the above reaction are very easily oxidised to carboxylc acids if allowed to remain in the reaction mixture. -

ISO Basic Latin Alphabet
ISO basic Latin alphabet The ISO basic Latin alphabet is a Latin-script alphabet and consists of two sets of 26 letters, codified in[1] various national and international standards and used widely in international communication. The two sets contain the following 26 letters each:[1][2] ISO basic Latin alphabet Uppercase Latin A B C D E F G H I J K L M N O P Q R S T U V W X Y Z alphabet Lowercase Latin a b c d e f g h i j k l m n o p q r s t u v w x y z alphabet Contents History Terminology Name for Unicode block that contains all letters Names for the two subsets Names for the letters Timeline for encoding standards Timeline for widely used computer codes supporting the alphabet Representation Usage Alphabets containing the same set of letters Column numbering See also References History By the 1960s it became apparent to thecomputer and telecommunications industries in the First World that a non-proprietary method of encoding characters was needed. The International Organization for Standardization (ISO) encapsulated the Latin script in their (ISO/IEC 646) 7-bit character-encoding standard. To achieve widespread acceptance, this encapsulation was based on popular usage. The standard was based on the already published American Standard Code for Information Interchange, better known as ASCII, which included in the character set the 26 × 2 letters of the English alphabet. Later standards issued by the ISO, for example ISO/IEC 8859 (8-bit character encoding) and ISO/IEC 10646 (Unicode Latin), have continued to define the 26 × 2 letters of the English alphabet as the basic Latin script with extensions to handle other letters in other languages.[1] Terminology Name for Unicode block that contains all letters The Unicode block that contains the alphabet is called "C0 Controls and Basic Latin". -

Esters Introduction Structurally, an Ester Is a Compound That Has an Alkoxy (OR) Group Attached to the Carbonyl Group
Esters Introduction Structurally, an ester is a compound that has an alkoxy (OR) group attached to the carbonyl group. O R C O R' R may be H, alkyl or aryl, while R’ may be alkyl or aryl only. Esters are widespread in nature. Many of the fragrances of flowers and fruits are due to the esters present. Ethyl butanoate is the chief component that accounts for the pineapple-like aroma and flavour of pineapples. 1:18 PM 1 Nomenclature of Esters Names of esters consist of two words that reflect the composite structure of the ester. The first word is derived from the alkyl group of the alcohol component, and the second word from the carboxylate group of the carboxylic acid component of the ester. The name of the carboxylate portion is derived by substituting the -ic acid suffix of the parent carboxylic acid with the –ate suffix. The alkyl group is cited first followed by the carboxylate group separated by a space. An ester is thus named as an alkyl 1:18 PM alkanoate. 2 IUPAC Nomenclature of Esters Examples 1:18 PM 3 Synthesis of Esters Preparative Strategies Highlighted below are some of the most common strategies by which esters are prepared. The esters are commonly prepared from the reaction of carboxylic acids, acid chlorides and acid anhydrides with alcohols. 1:18 PM 4 Synthesis of Esters Acid-Catalysed Esterification of a Carboxylic Acid and an Alcohol The acid-catalysed reaction of carboxylic acids and alcohols provides esters. Typically, a catalytic amount of a strong inorganic (mineral) acid such as H2SO4, HCl and H3PO4 is used. -

Stereochemistry, Alkyl Halide Substitution (SN1 & SN2)
Chem 261 Assignment & Lecture Outline 3: Stereochemistry, Alkyl Halide Substitution (SN1 & SN2) Read Organic Chemistry, Solomons, Fryle & Snyder 12th Edition (Electronic) • Functional Group List – Learn to recognize – Please see Green Handout – also p 76 of text • Periodic Table – Inside Front Cover - know 1st 10 elements (up through Neon) • Relative Strength of Acids and Bases – Inside Front cover (reference only) • Chapter 5 – Stereochemistry • Chapter 6 –Ionic Reactions: Nucleophilic Substitution of Alkyl Halides Problems: Do Not turn in, answers available in "Study Guide Student Solutions Manual " Solomons, Fryle, Snyder • Chapter 5: 5.1 to 5.15; 5.18 to 5.21; 5.26; 5.28; 5.33a-d; 5.46 • Chapter 6: 6.1 to 6.5; 6.7 to 6.10; 6.13; 6.20; 6.26; 6.27 Lecture Outline # 3 I. Comparison of 2 Structures: Same Molecular Formula ? -> If Yes, Possibly Isomers or Identical Same Arrangement (Sequence) of Groups ? If No -> Structural Isomers If Yes -> Superposable? If Yes -> Identical Structures If No -> Stereoisomers Non-Superposable Mirror Images ? If NO -> Diastereomers If Yes -> Enantiomers II. Chirality and Stereoisomers A. The Concept of Chirality 1. Identification of chiral objects a) achiral = not chiral b) planes of symmetry within a molecule 2. Types of stereoisomers – enantiomers and diastereomers B. Location of stereogenic (chiral) centres – 4 different groups on tetrahedral atom 1. Enantiomers & diastereomers 2. Meso compounds - chiral centers with plane of symmetry within molecule 3. Molecules with more than one chiral centre 4. Recognition of chiral centers in complex molecules - cholesterol - 8 chiral centres Drawing the enantiomer of cholesterol and its potential 255 stereoisomers 5. -

Hydrogen Atom Transfer-Mediated Cyclisations of Nitriles
Hydrogen Atom Transfer-Mediated Cyclisations of Nitriles Oliver J. Turner,*[a,b] John A. Murphy,*[b] David. J. Hirst[a] and Eric P. A. Talbot*[a]† Abstract: Hydrogen atom transfer-mediated intramolecular C-C coupling reactions between alkenes and nitriles, using PhSiH3 and catalytic Fe(acac)3, are described. This introduces a new strategic bond disconnection for ring-closing reactions, forming ketones via imine intermediates. Of note is the scope of the reaction, including formation of sterically hindered ketones, spirocycles and fused cyclic systems. In the early 1960s, Kwiatek and Seyler first reported the use of metal hydrides as catalysts in the hydrogenation of α,β- unsaturated compounds.[1,2] The discovery by Halpern,[3] later elegantly developed by Norton,[4] that metal-hydride hydrogen atom transfer (HAT) proceeded by a free-radical mechanism opened the door to a wide range of alkene hydrofunctionalisation reactions. But it was the pioneering work by Mukaiyama[5] on the catalytic hydration of alkenes, using Co(acac)2 and oxygen, that sparked wider interest in the field of alkene hydrofunctionalisation. As a result, there now exists an extensive ‘toolkit’ for the addition of hydrogen and a functional group to an alkene with Markovnikov selectivity and high chemo-selectivity using cobalt, manganese and iron complexes.[6,7] Efforts have also been made to extend HAT methodologies to C-C bond formation, both in an intra- and intermolecular fashion: Baran’s group developed a general C-C coupling reaction, utilising electron-deficient alkenes as capable radical acceptors (Scheme 1ai).[8–10] Hydropyridylation of alkenes by intramolecular Minisci reaction was recently demonstrated by Starr,[11] which allows for the formation of structures such as Scheme 1. -

Fermentation and Ester Taints
Fermentation and Ester Taints Anita Oberholster Introduction: Aroma Compounds • Grape‐derived –provide varietal distinction • Yeast and fermentation‐derived – Esters – Higher alcohols – Carbonyls – Volatile acids – Volatile phenols – Sulfur compounds What is and Esters? • Volatile molecule • Characteristic fruity and floral aromas • Esters are formed when an alcohol and acid react with each other • Few esters formed in grapes • Esters in wine ‐ two origins: – Enzymatic esterification during fermentation – Chemical esterification during long‐term storage Ester Formation • Esters can by formed enzymatically by both the plant and microbes • Microbes – Yeast (Non‐Saccharomyces and Saccharomyces yeast) – Lactic acid bacteria – Acetic acid bacteria • But mainly produced by yeast (through lipid and acetyl‐CoA metabolism) Ester Formation Alcohol function Keto acid‐Coenzyme A Ester Ester Classes • Two main groups – Ethyl esters – Acetate esters • Ethyl esters = EtOH + acid • Acetate esters = acetate (derivative of acetic acid) + EtOH or complex alcohol from amino acid metabolism Ester Classes • Acetate esters – Ethyl acetate (solvent‐like aroma) – Isoamyl acetate (banana aroma) – Isobutyl acetate (fruit aroma) – Phenyl ethyl acetate (roses, honey) • Ethyl esters – Ethyl hexanoate (aniseed, apple‐like) – Ethyl octanoate (sour apple aroma) Acetate Ester Formation • 2 Main factors influence acetate ester formation – Concentration of two substrates acetyl‐CoA and fusel alcohol – Activity of enzyme responsible for formation and break down reactions • Enzyme activity influenced by fermentation variables – Yeast – Composition of fermentation medium – Fermentation conditions Acetate/Ethyl Ester Formation – Fermentation composition and conditions • Total sugar content and optimal N2 amount pos. influence • Amount of unsaturated fatty acids and O2 neg. influence • Ethyl ester formation – 1 Main factor • Conc. of precursors – Enzyme activity smaller role • Higher fermentation temp formation • C and N increase small effect Saerens et al. -

Chapter 14 – Aldehydes and Ketones
Chapter 14 – Aldehydes and Ketones 14.1 Structures and Physical Properties of Aldehydes and Ketones Ketones and aldehydes are related in that they each possess a C=O (carbonyl) group. They differ in that the carbonyl carbon in ketones is bound to two carbon atoms (RCOR’), while that in aldehydes is bound to at least one hydrogen (H2CO and RCHO). Thus aldehydes always place the carbonyl group on a terminal (end) carbon, while the carbonyl group in ketones is always internal. Some common examples include (common name in parentheses): O O H HH methanal (formaldehyde) trans-3-phenyl-2-propenal (cinnamaldehyde) preservative oil of cinnamon O O propanone (acetone) 3-methylcyclopentadecanone (muscone) nail polish remover a component of one type of musk oil Simple aldehydes (e.g. formaldehyde) typically have an unpleasant, irritating odor. Aldehydes adjacent to a string of double bonds (e.g. 3-phenyl-2-propenal) frequently have pleasant odors. Other examples include the primary flavoring agents in oil of bitter almond (Ph- CHO) and vanilla (C6H3(OH)(OCH3)(CHO)). As your book says, simple ketones have distinctive odors (similar to acetone) that are typically not unpleasant in low doses. Like aldehydes, placing a collection of double bonds adjacent to a ketone carbonyl generally makes the substance more fragrant. The primary flavoring agent in oil of caraway is just a such a ketone. 2 O oil of carraway Because the C=O group is polar, small aldehydes and ketones enjoy significant water solubility. They are also quite soluble in typical organic solvents. 14.2 Naming Aldehydes and Ketones Aldehydes The IUPAC names for aldehydes are obtained by using rules similar to those we’ve seen for other functional groups (e.g.