208159Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
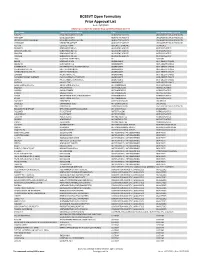
BCBSVT Open Formulary Prior Approval List
BCBSVT Open Formulary Prior Approval List As of: 10/27/2020 Helpful Tip: To search for a specific drug, use the find feature (Ctrl + F) Trade Name Chemical/Biological Name Class Prior Authorization Program FUSILEV LEVOLEUCOVORIN CALCIUM ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS KHAPZORY LEVOLEUCOVORIN ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS LEVOLEUCOVORIN CALCIUM LEVOLEUCOVORIN CALCIUM ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS VISTOGARD URIDINE TRIACETATE ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS ACTHAR CORTICOTROPIN ADRENAL HORMONES HORMONES BELRAPZO BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS BENDAMUSTINE HCL BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS BENDEKA BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS TREANDA BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS DAW (DISPENSE AS WRITTEN) ALL CUSTOM BELVIQ LORCASERIN HCL ANOREXIANTS ANTI‐OBESITY DRUGS BELVIQ XR LORCASERIN HCL ANOREXIANTS ANTI‐OBESITY DRUGS CONTRAVE ER NALTREXONE HCL/BUPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS DIETHYLPROPION HCL DIETHYLPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS DIETHYLPROPION HCL ER DIETHYLPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS LOMAIRA PHENTERMINE HCL ANOREXIANTS ANTI‐OBESITY DRUGS PHENDIMETRAZINE TARTRATE PHENDIMETRAZINE TARTRATE ANOREXIANTS ANTI‐OBESITY DRUGS QSYMIA PHENTERMINE/TOPIRAMATE ANOREXIANTS ANTI‐OBESITY DRUGS SAXENDA LIRAGLUTIDE ANOREXIANTS ANTI‐OBESITY DRUGS ABIRATERONE ACETATE ABIRATERONE ACETATE ANTIANDROGENS ANTINEOPLASTICS ERLEADA APALUTAMIDE ANTIANDROGENS ANTINEOPLASTICS NUBEQA DAROLUTAMIDE ANTIANDROGENS -

2018-2019 Targeted Medication Safety Best Practices for Hospitals
2018-2019 Targeted Medication Safety Best Practices for Hospitals The purpose of the Targeted Medication Safety Best Practices for Hospitals is to identify, inspire, and mobilize widespread, national adoption of consensus-based best practices for specific medication safety issues that continue to cause fatal and harmful errors in patients, despite repeated warnings in ISMP publications. Hospitals can focus their medication safety efforts over the next 2 years on these best practices, which are realistic and have been successfully adopted by numerous organizations. While targeted for the hospital-based setting, some best practices may be applicable to other healthcare settings. The Targeted Medication Safety Best Practices for Hospitals have been reviewed by an external expert advisory panel and approved by the ISMP Board of Trustees. Related issues of the ISMP Medication Safety Alert! are referenced after each best practice. ISMP encourages hospitals that have not implemented the 2016-2017 Targeted Medication Safety Best Practices for Hospitals to do so as a priority, while implementing the 2018-2019 best practices. Organizations need to focus on previous best practices 2, 3, 9 and 11 since these have the lowest implementation rate. Two of the 2016-2017 Targeted Medication Safety Best Practices for Hospitals (number 4 and 7) have been revised for 2018-2019. Best practices number 12 through 14 are new for 2018-2019. www.ismp.org BEST PRACTICE 1: Dispense vinCRIStine (and other vinca alkaloids) in a minibag of a compatible solution and not in a syringe. Rationale: Related ISMP Medication The goal of this best practice is to ensure that vinca alkaloids are Safety Alerts!: administered by the intravenous route only. -

Wednesday, February 10, 2016 4 P.M
Wednesday, February 10, 2016 4 p.m. Oklahoma Health Care Authority 4345 N. Lincoln Blvd. Oklahoma City, OK 73105 The University of Oklahoma Health Sciences Center COLLEGE OF PHARMACY PHARMACY MANAGEMENT CONSULTANTS MEMORANDUM TO: Drug Utilization Review (DUR) Board Members FROM: Bethany Holderread, Pharm.D. SUBJECT: Packet Contents for DUR Board Meeting – February 10, 2016 DATE: February 1, 2016 Note: The DUR Board will meet at 4:00p.m. The meeting will be held at 4345 N Lincoln Blvd. Enclosed are the following items related to the February meeting. Material is arranged in order of the agenda. Call to Order Public Comment Forum Action Item – Approval of DUR Board Meeting Minutes – Appendix A Update on Medication Coverage Authorization Unit/Oral Viscous Lidocaine Claims Analysis Update – Appendix B Action Item – Vote to Prior Authorize Duopa™ (Carbidopa/Levodopa Enteral Suspension) and Rytary™ (Carbidopa/Levodopa Extended-Release Capsules) – Appendix C Action Item – Vote to Prior Authorize Cortisporin® and Pediotic® (Neomycin/Polymyxin B/Hydrocortisone Otic) – Appendix D Action Item – Vote to Prior Authorize Migranal® (Dihydroergotamine Nasal Spray) – Appendix E Action Item – Vote to Prior Authorize Strensiq™ (Asfotase Alfa) – Appendix F Action Item – Vote to Prior Authorize Varubi™ (Rolapitant) – Appendix G Action Item – Vote to Prior Authorize Xuriden™ (Uridine Triacetate) – Appendix H Annual Review of Gout Medications and 30-Day Notice to Prior Authorize Mitigare™ (Colchicine Capsules) and Zurampic® (Lesinurad) – Appendix I Annual Review of Seizure Medications and 30-Day Notice to Prior Authorize Spritam® (Levetiracetam) – Appendix J 30-Day Notice to Prior Authorize Solaraze® (Diclofenac Gel) – Appendix K Annual Review of Ulcerative Colitis Medications and 30-Day Notice to Prior Authorize Uceris® (Budesonide Extended-Release Tablets), Uceris® (Budesonide Rectal Foam), and Miscellaneous Mesalamine Products – Appendix L Annual Review of Ocular Allergy Medications and 30-Day Notice to Prior Authorize Pazeo® (Olopatadine Ophthalmic) – Appendix M ORI-4403 • P.O. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

Database for Drug Metabolism and Comparisons, Nicedrug.Ch, Aids
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.28.120782; this version posted June 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Database for drug metabolism and comparisons, NICEdrug.ch, aids discovery and design 2 3 Authors/affiliation 1 1,2,5 1,3,5 4 Homa MohammadiPeyhani , Anush Chiappino-Pepe , Kiandokht Haddadi , Jasmin 1 1,4 1,* 5 Hafner , Noushin Hadadi , Vassily Hatzimanikatis 6 1 7 Laboratory of Computational Systems Biotechnology, École Polytechnique Fédérale de 8 Lausanne, EPFL, Lausanne, Switzerland 2 9 Present address: Department of Genetics, Harvard Medical School, Boston, Massachusetts, 10 USA 3 11 Present address: Department of Chemical Engineering and Applied Chemistry, University of 12 Toronto, Toronto, Canada 4 13 Present address: Department of Cell Physiology and Metabolism, Université de Genève, 14 Geneva, Switzerland 5 15 These authors contributed equally 16 * 17 Corresponding author: 18 Prof. Vassily Hatzimanikatis 19 Laboratory of Computational Systems Biotechnology (LCSB), École Polytechnique Fédérale de 20 Lausanne (EPFL), CH‐1015 21 Lausanne, Switzerland 22 Email: [email protected] , Phone: +41 (0)21 693 98 70, Fax: +41 (0)21 693 98 75 23 24 Abstract 25 The discovery of a drug requires over a decade of enormous research and financial 26 investments—and still has a high risk of failure. To reduce this burden, we developed the 27 NICEdrug.ch database, which incorporates 250,000 bio-active molecules, and studied their 28 metabolic targets, fate, and toxicity. -

New Drug Approvals and Extended Indications for Infants, Children, and Adolescents Marcia L
PEDIATRIC PHARMACOTHERAPY Volume 21 Number 11 November 2015 New Drug Approvals and Extended Indications for Infants, Children, and Adolescents Marcia L. Buck, PharmD, FCCP, FPPAG ver the past six months, a number of which slowly releases the drug over time, O significant new drugs have been approved providing up to 13 hours of symptom control. by the Food and Drug Administration (FDA). In The safety and efficacy of the product was addition, several drugs already on the market established in a phase 3 randomized, placebo- have been granted an indication for use in controlled trial in 108 children with ADHD.4 pediatric patients. Following a 5-week open-label dose optimization period, patients were randomized to New Drug Product Approvals treatment (2.5-10 mg) or placebo for a 1-week period. At the end of the week, scores on the Adapalene and Benzoyl Peroxide Swanson, Kotkin, Agler, M-Flynn, and Pelham Epiduo Forte®, a new product containing (SKAMP)-Combined rating scale were adapalene 0.3%, a retinoid, and benzoyl peroxide compared to baseline. The change in scores after 2.5% was approved on July 16, 2015 for the treatment demonstrated a statistically significant treatment of moderate to severe acne in adults improvement throughout the day compared to and children 12 years of age and older.1 The placebo (assessed at 1, 2, 6, 8, 10, 12, and 13 efficacy of the new product was demonstrated in hours post-dose), with a mean change of -8.8 (SE a phase 3 multicenter randomized, double-blind 1.14) in the treated patients and 6.0 (SE 1.19) in trial comparing it to the gel vehicle without the the controls. -

Specialty Pharmacy Program Drug List
Specialty Pharmacy Program Drug List The Specialty Pharmacy Program covers certain drugs commonly referred to as high-cost Specialty Drugs. To receive in- network benefits/coverage for these drugs, these drugs must be dispensed through a select group of contracted specialty pharmacies or your medical provider. Please call the BCBSLA Customer Service number on the back of your member ID card for information about our contracted specialty pharmacies. All specialty drugs listed below are limited to the retail day supply listed in your benefit plan (typically a 30-day supply). As benefits may vary by group and individual plans, the inclusion of a medication on this list does not imply prescription drug coverage. Please refer to your benefit plan for a complete list of benefits, including specific exclusions, limitations and member cost-sharing amounts you are responsible for such as a deductible, copayment and coinsurance. Brand Name Generic Name Drug Classification 8-MOP methoxsalen Psoralen ACTEMRA SC tocilizumab Monoclonal Antibody/Arthritis ACTHAR corticotropin Adrenocortical Insufficiency ACTIMMUNE interferon gamma 1b Interferon ADCIRCA tadalafil Pulmonary Vasodilator ADEMPAS riociguat Pulmonary Vasodilator AFINITOR everolimus Oncology ALECENSA alectinib Oncology ALKERAN (oral) melphalan Oncology ALUNBRIG brigatinib Oncology AMPYRA ER dalfampridine Multiple Sclerosis APTIVUS tipranavir HIV/AIDS APOKYN apomorphine Parkinson's Disease ARCALYST rilonacept Interleukin Blocker/CAPS ATRIPLA efavirenz-emtricitabine-tenofovir HIV/AIDS AUBAGIO -

Management of 5-Fluorouracil (5FU) Infusion Overdose at BC Cancer
BC Cancer Agency Management Guidelines Management of 5-fluorouracil (5FU) infusion overdose Rationale: 5-FU is an analog of uracil and acts as a pyrimidine antagonist. It is widely used to treat solid tumors and is often administered via infusion devices at or near its maximum tolerated dose. The common use of infusion devices containing 5FU for infusion over several days increases the possibility of potentially lethal overdoses due to device malfunction, dose calculation errors, device selection errors or misprogramming. Toxicities resulting from delivery of 5FU at greater than the intended dose or dose rate can range from mild to life-threatening depending upon the rate of infusion and total dose delivered. Presently, no standard guidelines exist regarding the definition of a 5FU infusion overdose and the recommended management. This guidance serves as a directive regarding the overdose management for 5FU infusors at the BC Cancer Agency (BCCA). More detailed management information can be found in POISINDEX management of fluorouracil and related agents. Directive: Prescribing medical staff, pharmacy and nursing must ensure that every effort is made to minimize the risk of an error in dose calculation or infusor misprogramming. In the event of a 5FU overdose, appropriate and timely measures to anticipate and implement supportive management of patients at risk for severe 5FU toxicity should be implemented per the procedures outlines below. Anticipated toxicities of 5FU can include but are not limited to myelosuppression, nausea, vomiting, stomatitis, diarrhea, ileus, and ileitis. Less common but severe effects can include hepatotoxicity, cardiogenic pulmonary edema, seizures, shock, gastrointestinal bleeding and perforation. Procedures: For the purposes of this guidance, a 5FU infusor overdose will be empirically defined as administration of 5FU via infusor at greater than or equal to 2 times the intended rate with completed delivery of greater than 50% of the intended total 5FU dose. -

Uridine Triacetate
PATIENT & CAREGIVER EDUCATION Uridine Triacetate This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Vistogard; Xuriden What is this drug used for? It is used to treat a rare metabolism health problem called orotic aciduria. It is used to treat fluorouracil or capecitabine overdose. It is used to treat very bad or life-threatening side effects caused by fluorouracil or capecitabine. What do I need to tell my doctor BEFORE I take this drug? If you are allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell your doctor about the allergy and what signs you had. This drug may interact with other drugs or health problems. Tell your doctor and pharmacist about all of your drugs (prescription or OTC, natural products, vitamins) and health problems. You must check to make sure that it is safe for you to take this drug with all of your drugs and health problems. Do not start, stop, or change the dose of any drug without checking with your doctor. Uridine Triacetate 1/6 What are some things I need to know or do while I take this drug? For all uses of this drug: Tell all of your health care providers that you take this drug. This includes your doctors, nurses, pharmacists, and dentists. Have blood work checked as you have been told by the doctor. Talk with the doctor. -

Essential Medications Review
Essential medications for high-quality patient care Quarter 3 2021 update Essential medications – September 2021 As part of the mission to end drug shortages, the Vizient team of pharmacy experts continues to identify essential medications where, if not available, would prove the greatest threat to hospitals’ ability to provide immediate and high-quality patient care. Medications identified as of greatest importance were selected by the Vizient pharmacy team from a comprehensive clinical review of products contained within the World Health Organization’s (WHO) Essential Medicines list, the Advanced Cardiac Life Support (ACLS) and Pediatric Advanced Life Support (PALS) algorithms, and medications included in Vizient member health systems’ critical drug lists. As of this edition, 14 drugs were added to the list creating a total of 251 line items, representing 237 unique drugs and three categories. This includes: • Acute treatment drugs with no alternatives (77 drugs) – Medicines used in acute and critical circumstances to sustain life and for which there are no current alternatives • Chronic treatment drugs with no alternatives (37 drugs) – Products used in chronic disease states or conditions where no alternatives are available (e.g., chemotherapy medications) • High impact drugs (137 drugs) – Medicines for which alternatives are available but may be less clinically desirable and/or are more operationally difficult to use; also reflects drugs where the absence of one medication can affect therapeutically related drugs. Updated quarterly, -

ENTRY WATCH 2016 Published by the Patented Medicine Prices Review Board June 2018 Meds Entry Watch, 2016 Is Available in Electronic Format on the PMPRB Website
MEDS ENTRY WATCH 2016 Published by the Patented Medicine Prices Review Board June 2018 Meds Entry Watch, 2016 is available in electronic format on the PMPRB website. Une traduction de ce document est également disponible en français sous le titre : Veille des médicaments mis en marché, 2016 Patented Medicine Prices Review Board Standard Life Centre Box L40 333 Laurier Avenue West Suite 1400 Ottawa, ON K1P 1C1 Tel.: 1-877-861-2350 TTY 613-288-9654 Email: [email protected] Web: www.pmprb-cepmb.gc.ca ISSN 2560-6204 Cat. No.: H79-12E-PDF © Her Majesty the Queen in Right of Canada, as represented by the NPDUIS initiative of the Patented Medicine Prices Review Board, 2018 MEDS ENTRY WATCH 2016 About the PMPRB Acknowledgements The Patented Medicine Prices Review Board This report was prepared by the Patented (PMPRB) is a respected public agency that makes Medicine Prices Review Board (PMPRB) a unique and valued contribution to sustainable as part of the National Prescription Drug spending on pharmaceuticals in Canada by: Utilization Information System (NPDUIS). ~ providing stakeholders with price, cost and The PMPRB would like to acknowledge the utilization information to help them make timely contributions of and knowledgeable drug pricing, purchasing and ~ The members of the NPDUIS Advisory reimbursement decisions; and Committee for their expert oversight and ~ acting as an effective check on the patent rights guidance in the preparation of this report. of pharmaceutical manufacturers through the ~ PMPRB NPDUIS staff for their contribution responsible and efficient use of its consumer to the analytical content of the report: protection powers. -

Prior Authorization Approval Criteria
Prior Authorization Approval Criteria Effective Date: 10/01/2020 Standard Commercial Prior Authorization Guidelines STANDARD COMMERCIAL DRUG FORMULARY PRIOR AUTHORIZATION GUIDELINES 1. Formulary Agents Drug products that are listed in the Formulary as Prior Authorization (PA) require evaluation, per MedImpact Pharmacy and Therapeutics Committee guidelines, when the member presents a prescription to a network pharmacy. Each request will be reviewed on individual patient need. If the request does not meet the criteria established by the P & T Committee, the request will not be approved and alternative therapy will be recommended. 2. Non-Formulary Agents Any product not found in the Formulary listing, or any Formulary updates published by MedImpact, shall be considered a Non-Formulary drug. Coverage for non-formulary agents may be applied for in advance. When a member gives a prescription order for a non-formulary drug to a pharmacist, the pharmacist will evaluate the patient’s drug history and contact the physician to determine if there is a legitimate medical need for a non-formulary drug. Each request will be reviewed on individual patient need. The following basic criteria are used: a. The use of Formulary Drug Products is contraindicated in the patient. b. The patient has failed an appropriate trial of Formulary or related agents. c. The choices available in the Drug Formulary are not suited for the present patient care need, and the drug selected is required for patient safety. d. The use of a Formulary drug may provoke an underlying condition, which would be detrimental to patient care. If the request does not meet the criteria established by the P & T Committee, the request will not be approved and alternative therapy will be recommended.