6. ANALYTICAL METHODS the Purpose of This Chapter Is To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Emerging Technologies for Degradation of Dichlorvos: a Review
International Journal of Environmental Research and Public Health Review Emerging Technologies for Degradation of Dichlorvos: A Review Yuming Zhang 1,2, Wenping Zhang 1,2, Jiayi Li 1,2, Shimei Pang 1,2, Sandhya Mishra 1,2 , Pankaj Bhatt 1,2 , Daxing Zeng 3,* and Shaohua Chen 1,2,* 1 State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangdong Province Key Laboratory of Microbial Signals and Disease Control, Integrative Microbiology Research Centre, South China Agricultural University, Guangzhou 510642, China; [email protected] (Y.Z.); [email protected] (W.Z.); [email protected] (J.L.); [email protected] (S.P.); [email protected] (S.M.); [email protected] (P.B.) 2 Guangdong Laboratory for Lingnan Modern Agriculture, Guangzhou 510642, China 3 School of Applied Chemistry and Biological Technology, Shenzhen Polytechnic, Shenzhen 518055, China * Correspondence: [email protected] (D.Z.); [email protected] (S.C.); Tel.: +86-20-8528 8229 (S.C.) Abstract: Dichlorvos (O,O-dimethyl O-(2,2-dichlorovinyl)phosphate, DDVP) is a widely acknowl- edged broad-spectrum organophosphorus insecticide and acaracide. This pesticide has been used for more than four decades and is still in strong demand in many developing countries. Extensive application of DDVP in agriculture has caused severe hazardous impacts on living systems. The International Agency for Research on Cancer of the World Health Organization considered DDVP among the list of 2B carcinogens, which means a certain extent of cancer risk. Hence, removing DDVP from the environment has attracted worldwide attention. Many studies have tested the removal of DDVP using different kinds of physicochemical methods including gas phase surface discharge plasma, physical adsorption, hydrodynamic cavitation, and nanoparticles. -

Nerve Agent - Lntellipedia Page 1 Of9 Doc ID : 6637155 (U) Nerve Agent
This document is made available through the declassification efforts and research of John Greenewald, Jr., creator of: The Black Vault The Black Vault is the largest online Freedom of Information Act (FOIA) document clearinghouse in the world. The research efforts here are responsible for the declassification of MILLIONS of pages released by the U.S. Government & Military. Discover the Truth at: http://www.theblackvault.com Nerve Agent - lntellipedia Page 1 of9 Doc ID : 6637155 (U) Nerve Agent UNCLASSIFIED From lntellipedia Nerve Agents (also known as nerve gases, though these chemicals are liquid at room temperature) are a class of phosphorus-containing organic chemicals (organophosphates) that disrupt the mechanism by which nerves transfer messages to organs. The disruption is caused by blocking acetylcholinesterase, an enzyme that normally relaxes the activity of acetylcholine, a neurotransmitter. ...--------- --- -·---- - --- -·-- --- --- Contents • 1 Overview • 2 Biological Effects • 2.1 Mechanism of Action • 2.2 Antidotes • 3 Classes • 3.1 G-Series • 3.2 V-Series • 3.3 Novichok Agents • 3.4 Insecticides • 4 History • 4.1 The Discovery ofNerve Agents • 4.2 The Nazi Mass Production ofTabun • 4.3 Nerve Agents in Nazi Germany • 4.4 The Secret Gets Out • 4.5 Since World War II • 4.6 Ocean Disposal of Chemical Weapons • 5 Popular Culture • 6 References and External Links --------------- ----·-- - Overview As chemical weapons, they are classified as weapons of mass destruction by the United Nations according to UN Resolution 687, and their production and stockpiling was outlawed by the Chemical Weapons Convention of 1993; the Chemical Weapons Convention officially took effect on April 291997. Poisoning by a nerve agent leads to contraction of pupils, profuse salivation, convulsions, involuntary urination and defecation, and eventual death by asphyxiation as control is lost over respiratory muscles. -

Chlorpyrifos (Dursban) Ddvp (Dichlorvos) Diazinon Malathion Parathion
CHLORPYRIFOS (DURSBAN) DDVP (DICHLORVOS) DIAZINON MALATHION PARATHION Method no.: 62 Matrix: Air Procedure: Samples are collected by drawing known volumes of air through specially constructed glass sampling tubes, each containing a glass fiber filter and two sections of XAD-2 adsorbent. Samples are desorbed with toluene and analyzed by GC using a flame photometric detector (FPD). Recommended air volume and sampling rate: 480 L at 1.0 L/min except for Malathion 60 L at 1.0 L/min for Malathion Target concentrations: 1.0 mg/m3 (0.111 ppm) for Dichlorvos (PEL) 0.1 mg/m3 (0.008 ppm) for Diazinon (TLV) 0.2 mg/m3 (0.014 ppm) for Chlorpyrifos (TLV) 15.0 mg/m3 (1.11 ppm) for Malathion (PEL) 0.1 mg/m3 (0.008 ppm) for Parathion (PEL) Reliable quantitation limits: 0.0019 mg/m3 (0.21 ppb) for Dichlorvos (based on the RAV) 0.0030 mg/m3 (0.24 ppb) for Diazinon 0.0033 mg/m3 (0.23 ppb) for Chlorpyrifos 0.0303 mg/m3 (2.2 ppb) for Malathion 0.0031 mg/m3 (0.26 ppb) for Parathion Standard errors of estimate at the target concentration: 5.3% for Dichlorvos (Section 4.6.) 5.3% for Diazinon 5.3% for Chlorpyrifos 5.6% for Malathion 5.3% for Parathion Status of method: Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. Date: October 1986 Chemist: Donald Burright Organic Methods Evaluation Branch OSHA Analytical Laboratory Salt Lake City, Utah 1 of 27 T-62-FV-01-8610-M 1. -

Dichlorvos in Drinking-Water
WHO/FWC/WSH/16.44 Dichlorvos in Drinking-water Background document for development of WHO Guidelines for Drinking-water Quality World Health Organization 2016 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; email: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for non- commercial distribution – should be addressed to WHO Press at the above address (fax: +41 22791 4806; email: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by the World Health Organization to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either expressed or implied. The responsibility for the interpretation and use of the material lies with the reader. -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Lifetime Organophosphorous Insecticide Use Among Private Pesticide Applicators in the Agricultural Health Study
Journal of Exposure Science and Environmental Epidemiology (2012) 22, 584 -- 592 & 2012 Nature America, Inc. All rights reserved 1559-0631/12 www.nature.com/jes ORIGINAL ARTICLE Lifetime organophosphorous insecticide use among private pesticide applicators in the Agricultural Health Study Jane A. Hoppin1, Stuart Long2, David M. Umbach3, Jay H. Lubin4, Sarah E. Starks5, Fred Gerr5, Kent Thomas6, Cynthia J. Hines7, Scott Weichenthal8, Freya Kamel1, Stella Koutros9, Michael Alavanja9, Laura E. Beane Freeman9 and Dale P. Sandler1 Organophosphorous insecticides (OPs) are the most commonly used insecticides in US agriculture, but little information is available regarding specific OP use by individual farmers. We describe OP use for licensed private pesticide applicators from Iowa and North Carolina in the Agricultural Health Study (AHS) using lifetime pesticide use data from 701 randomly selected male participants collected at three time periods. Of 27 OPs studied, 20 were used by 41%. Overall, 95% had ever applied at least one OP. The median number of different OPs used was 4 (maximum ¼ 13). Malathion was the most commonly used OP (74%) followed by chlorpyrifos (54%). OP use declined over time. At the first interview (1993--1997), 68% of participants had applied OPs in the past year; by the last interview (2005--2007), only 42% had. Similarly, median annual application days of OPs declined from 13.5 to 6 days. Although OP use was common, the specific OPs used varied by state, time period, and individual. Much of the variability in OP use was associated with the choice of OP, rather than the frequency or duration of application. -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Current Awareness in Clinical Toxicology Editors: Damian Ballam Msc and Allister Vale MD
Current Awareness in Clinical Toxicology Editors: Damian Ballam MSc and Allister Vale MD October 2017 CONTENTS General Toxicology 11 Metals 31 Management 17 Pesticides 33 Drugs 19 Chemical Warfare 34 Chemical Incidents & 27 Plants 34 Pollution Chemicals 28 Animals 35 CURRENT AWARENESS PAPERS OF THE MONTH The interpretation of hair analysis for drugs and drug metabolites Cuypers E, Flanagan RJ. Clin Toxicol 2017; online early: doi: 10.1080/ 15563650.2017.1379603: Introduction Head hair analysis for drugs and drug metabolites has been used widely with the aim of detecting exposure in the weeks or months prior to sample collection. However, inappropriate interpretation of results has likely led to serious miscarriages of justice, especially in child custody cases. Objective The aim of this review is to assess critically what can, and perhaps more importantly, what cannot be claimed as regards the interpretation of hair test results in a given set of circumstances in order to inform future testing. Methods We searched the PubMed database for papers published 2010-2016 using the terms "hair" and "drug" and "decontamination", the terms "hair" and "drug" and "contamination", the terms "hair" and "drug-facilitated crime", the terms "hair" and "ethyl glucuronide", and the Current Awareness in Clinical Toxicology is produced monthly for the American Academy of Clinical Toxicology by the Birmingham Unit of the UK National Poisons Information Service, with contributions from the Cardiff, Edinburgh, and Newcastle Units. The NPIS is commissioned by Public Health England 2 terms "hair", "drug testing" and "analysis". Study of the reference lists of the 46 relevant papers identified 25 further relevant citations, giving a total of 71 citations. -

Environmental Health Criteria 79 Dichlorvos
Environmental Health Criteria 79 Dichlorvos Please note that the layout and pagination of this web version are not identical with the printed version. Dichlorvos (EHC 79, 1988) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 79 DICHLORVOS This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. First draft prepared by Dr. J. Sekizawa (National Institute of Hygienic Sciences, Japan) and Dr. M. Eto (Kyushu University, Japan) with the assistance of Dr. J. Miyamoto and Dr. M. Matsuo (Sumitomo Chemical Company) Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1989 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination of laboratory testing and epidemiological studies, and promotion of research on the mechanisms of the biological action of chemicals. ISBN 92 4 154279 9 Page 1 of 115 Dichlorvos (EHC 79, 1988) The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. -

Draft Human Health and Ecological Risk Assessment for Dichlorvos (DDVP) in Exotic Fruit Fly Applications
United States Department of Agriculture Draft Human Health and Marketing and Regulatory Ecological Risk Programs Animal and Plant Assessment for Dichlorvos Health Inspection Service (DDVP) in Exotic Fruit Fly Applications April 2018 Draft Human Health and Ecological Risk Assessment for Dichlorvos (DDVP) in Exotic Fruit Fly Applications April 2018 Agency Contact: John Stewart National Fruit Fly Policy Manager Plant Protection and Quarantine – Policy Management Animal and Plant Health Inspection Service U.S. Department of Agriculture 1730 Varsity Drive, Suite 400 Raleigh, NC 27606 i Non-Discrimination Policy The U.S. Department of Agriculture (USDA) prohibits discrimination against its customers, employees, and applicants for employment on the bases of race, color, national origin, age, disability, sex, gender identity, religion, reprisal, and where applicable, political beliefs, marital status, familial or parental status, sexual orientation, or all or part of an individual's income is derived from any public assistance program, or protected genetic information in employment or in any program or activity conducted or funded by the Department. (Not all prohibited bases will apply to all programs and/or employment activities.) To File an Employment Complaint If you wish to file an employment complaint, you must contact your agency's EEO Counselor (PDF) within 45 days of the date of the alleged discriminatory act, event, or in the case of a personnel action. Additional information can be found online at http://www.ascr.usda.gov/complaint_filing_file.html. To File a Program Complaint If you wish to file a Civil Rights program complaint of discrimination, complete the USDA Program Discrimination Complaint Form (PDF), found online at http://www.ascr.usda.gov/complaint_filing_cust.html, or at any USDA office, or call (866) 632- 9992 to request the form. -
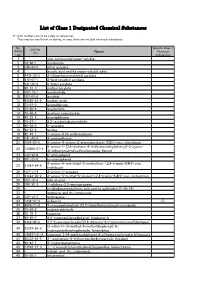
List of Class 1 Designated Chemical Substances
List of Class 1 Designated Chemical Substances *1:CAS numbers are to be solely as references. They may be insufficient or lacking, in case there are multiple chemical substances. No. Specific Class 1 CAS No. (PRTR Chemical (*1) Name Law) Substances 1 - zinc compounds(water-soluble) 2 79-06-1 acrylamide 3 140-88-5 ethyl acrylate 4 - acrylic acid and its water-soluble salts 5 2439-35-2 2-(dimethylamino)ethyl acrylate 6 818-61-1 2-hydroxyethyl acrylate 7 141-32-2 n-butyl acrylate 8 96-33-3 methyl acrylate 9 107-13-1 acrylonitrile 10 107-02-8 acrolein 11 26628-22-8 sodium azide 12 75-07-0 acetaldehyde 13 75-05-8 acetonitrile 14 75-86-5 acetone cyanohydrin 15 83-32-9 acenaphthene 16 78-67-1 2,2'-azobisisobutyronitrile 17 90-04-0 o-anisidine 18 62-53-3 aniline 19 82-45-1 1-amino-9,10-anthraquinone 20 141-43-5 2-aminoethanol 21 1698-60-8 5-amino-4-chloro-2-phenylpyridazin-3(2H)-one; chloridazon 5-amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-3-cyano- 22 120068-37-3 4[(trifluoromethyl)sulfinyl]pyrazole; fipronil 23 123-30-8 p-aminophenol 24 591-27-5 m-aminophenol 4-amino-6-tert-butyl-3-methylthio-1,2,4-triazin-5(4H)-one; 25 21087-64-9 metribuzin 26 107-11-9 3-amino-1-propene 27 41394-05-2 4-amino-3-methyl-6-phenyl-1,2,4-triazin-5(4H)-one; metamitron 28 107-18-6 allyl alcohol 29 106-92-3 1-allyloxy-2,3-epoxypropane 30 - n-alkylbenzenesulfonic acid and its salts(alkyl C=10-14) 31 - antimony and its compounds 32 120-12-7 anthracene 33 1332-21-4 asbestos ○ 34 4098-71-9 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate 35 78-84-2 isobutyraldehyde -

Alternatives to Dichlorvos After It Is Banned
Quality Controlling insects during storage Alternatives to dichlorvos after it is banned The new Maximum Residue Levels set by European Regulations for the use of two storage insecticides are so low that it is no longer possible to apply them directly to grain. There are already solutions to replace malathion, but none to replace dichlorvos. Here are details of the measures required to compensate for their withdrawal. European regulations on the use of insecticides in storage are becoming more stringent. The withdrawal of malathion and dichlorvos has been scheduled for 2008, but already their Maximum Residue Levels (MRL) have been reduced so significantly (0.5 ppm and 0.01 ppm respectively), that it is no longer possible to apply them directly to grain. There are already solutions to replace malathion, including using three molecules which are still permitted: chlorpyrifos-methyl, pirimiphos-methyl and deltamethrin. But there are no available alternatives for dichlorvos (DDVP), which was the only “shock treatment” insecticide to help treat grain batches infested with insects shortly before dispatch. Its disappearance will mean having to implement new measures from this autumn onwards. Grain cleaning is becoming a must Cleanliness of premises is essential Cleaning premises and equipment is foremost on the list of insect control measures, since it helps eradicate any sources of protection and food that insects need when silos are empty of grain. When facilities are not in operation, using compressed air is often enough to loosen dust deposited in the upper regions of the installation and make it fall to the ground. However, this is not effective if the premises were infested with moths, especially Plodia interpunctella.