Pan-Genome Analyses of 24 Shewanella Strains Re-Emphasize
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Gut Microbiome of the Sea Urchin, Lytechinus Variegatus, from Its Natural Habitat Demonstrates Selective Attributes of Micro
FEMS Microbiology Ecology, 92, 2016, fiw146 doi: 10.1093/femsec/fiw146 Advance Access Publication Date: 1 July 2016 Research Article RESEARCH ARTICLE The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles Joseph A. Hakim1,†, Hyunmin Koo1,†, Ranjit Kumar2, Elliot J. Lefkowitz2,3, Casey D. Morrow4, Mickie L. Powell1, Stephen A. Watts1,∗ and Asim K. Bej1,∗ 1Department of Biology, University of Alabama at Birmingham, 1300 University Blvd, Birmingham, AL 35294, USA, 2Center for Clinical and Translational Sciences, University of Alabama at Birmingham, Birmingham, AL 35294, USA, 3Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL 35294, USA and 4Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, 1918 University Blvd., Birmingham, AL 35294, USA ∗Corresponding authors: Department of Biology, University of Alabama at Birmingham, 1300 University Blvd, CH464, Birmingham, AL 35294-1170, USA. Tel: +1-(205)-934-8308; Fax: +1-(205)-975-6097; E-mail: [email protected]; [email protected] †These authors contributed equally to this work. One sentence summary: This study describes the distribution of microbiota, and their predicted functional attributes, in the gut ecosystem of sea urchin, Lytechinus variegatus, from its natural habitat of Gulf of Mexico. Editor: Julian Marchesi ABSTRACT In this paper, we describe the microbial composition and their predictive metabolic profile in the sea urchin Lytechinus variegatus gut ecosystem along with samples from its habitat by using NextGen amplicon sequencing and downstream bioinformatics analyses. The microbial communities of the gut tissue revealed a near-exclusive abundance of Campylobacteraceae, whereas the pharynx tissue consisted of Tenericutes, followed by Gamma-, Alpha- and Epsilonproteobacteria at approximately equal capacities. -

26Sor Em2rray /Mmucd
D4006- Gut Microbiota Analysis UC Davis MMPC - Microbiome & Host Response Core Contents 1 Methods: 1 1.1 Sequencing . .1 1.2 Data processing . .1 2 Summary of Findings: 2 2.1 Sequencing analysis . .2 2.2 Microbial diversity . .2 2.2.1 Alpha Diversity . .2 2.2.2 Beta Diversity . 10 2.3 Data analysis using taxa abundance data . 13 2.3.1 Stacked bar graphs of Taxa abundances at each level . 14 A Appendix 1 (Taxa Abundance Tables) 30 B Appendix 2 (Taxa removed from filtered datasets at each level) 37 References: 43 Core Contacts: Helen E. Raybould, Ph.D., Core Leader ([email protected]) Trina A. Knotts, Ph.D., Core Co-Leader ([email protected]) Michael L. Goodson, Ph.D., Core Scientist ([email protected]) Client(s): Kent Lloyd, DVM PhD ;MMRRC; UC Davis Project #: MBP-2079 MMRRC strain ID: MMRRC_043514 Animal Information: The strain was donated to the MMRRC by Russell Ray at Baylor College of Medicine. Fecal samples were obtained from animals housed under the care of Russell Ray at Baylor College of Medicine. 1 Methods: Brief Project Description: MMRRC strains are often contributed to the MMRRC to fulfill the resource sharing aspects of NIH grants. Since transporting mice to another facilty often causes a microbiota shift, having a record of the original fecal microbiota from the donor institution where the original phenotyping or testing was performed may prove helpful if a phenotype is lost after transfer. Several MMRRC mouse lines were selected for fecal microbiota profiling of the microbiota. Table 1: Animal-Strain Information X.SampleID TreatmentGroup Animal_ID Genotype Line Sex MMRRC.043514.Mx054 MMRRC.043514_Het_M Mx054 Het MMRRC.043514 M MMRRC.043514.Mx415 MMRRC.043514_Het_M Mx415 Het MMRRC.043514 M 1 1.1 Sequencing Frozen fecal or regional gut samples were shipped on dry ice to UC Davis MMPC and Host Microbe Systems Biology Core. -

Thalassomonas Agarivorans Sp. Nov., a Marine Agarolytic Bacterium Isolated from Shallow Coastal Water of An-Ping Harbour, Taiwan
International Journal of Systematic and Evolutionary Microbiology (2006), 56, 1245–1250 DOI 10.1099/ijs.0.64130-0 Thalassomonas agarivorans sp. nov., a marine agarolytic bacterium isolated from shallow coastal water of An-Ping Harbour, Taiwan, and emended description of the genus Thalassomonas Wen Dar Jean,1 Wung Yang Shieh2 and Tung Yen Liu2 Correspondence 1Center for General Education, Leader University, No. 188, Sec. 5, An-Chung Rd, Tainan, Wung Yang Shieh Taiwan [email protected] 2Institute of Oceanography, National Taiwan University, PO Box 23-13, Taipei, Taiwan A marine agarolytic bacterium, designated strain TMA1T, was isolated from a seawater sample collected in a shallow-water region of An-Ping Harbour, Taiwan. It was non-fermentative and Gram-negative. Cells grown in broth cultures were straight or curved rods, non-motile and non-flagellated. The isolate required NaCl for growth and exhibited optimal growth at 25 6C and 3 % NaCl. It grew aerobically and was incapable of anaerobic growth by fermenting glucose or other carbohydrates. Predominant cellular fatty acids were C16 : 0 (17?5 %), C17 : 1v8c (12?8 %), C17 : 0 (11?1 %), C15 : 0 iso 2-OH/C16 : 1v7c (8?6 %) and C13 : 0 (7?3 %). The DNA G+C content was 41?0 mol%. Phylogenetic, phenotypic and chemotaxonomic data accumulated in this study revealed that the isolate could be classified in a novel species of the genus Thalassomonas in the family Colwelliaceae. The name Thalassomonas agarivorans sp. nov. is proposed for the novel species, with TMA1T (=BCRC 17492T=JCM 13379T) as the type strain. Alteromonas-like bacteria in the class Gammaproteobacteria however, they are not exclusively autochthonous in the comprise a large group of marine, heterotrophic, polar- marine environment, since some reports have shown that flagellated, Gram-negative rods that are mainly non- they also occur in freshwater, sewage and soil (Agbo & Moss, fermentative aerobes. -

Dddy Is a Bacterial Dimethylsulfoniopropionate Lyase Representing a New Cupin Enzyme Superfamily with Unknown Primary Function
bioRxiv preprint doi: https://doi.org/10.1101/161257; this version posted July 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. DddY is a bacterial dimethylsulfoniopropionate lyase representing a new cupin enzyme superfamily with unknown primary function Lei Lei, Uria Alcolombri & Dan S Tawfik Department of Biomolecular Sciences, Weizmann Institute of Science, Rehovot 76100, Israel. * Correspondence to: [email protected] Abstract Dimethylsulfide (DMS) is released at rates of >107 tons annually and plays a key role in the oceanic sulfur cycle and ecology. Marine bacteria, algae, and possibly other organisms, release DMS via cleavage of dimethylsulfoniopropionate (DMSP). Different genes encoding proteins with DMSP lyase activity are known belonging to different superfamilies and exhibiting highly variable levels of DMSP lyase activity. DddY shows the highest activity among all reported bacterial lyases yet is poorly characterized. Here, we describe the characterization of recombinant DddY is from different marine bacteria. We found that DddY activity demands a transition metal ion cofactor. DddY also shares two sequence motifs with other bacterial lyases assigned as cupin-like enzymes, DddQ, DddL, DddK, and DddW. These cupin motif residues are essential for DddY activity, as for the other cupin DMSP lyases, and all these enzymes are characterized by a common metal- chelator inhibitor (TPEN). Analysis of all sequences carrying these cupin motifs defined a superfamily: Cupin-DLL (DMSP lyases and lyase-like). The DMSP lyase families are sporadically distributed suggesting that DMSP lyases evolved within this superfamily independently along multiple lineages. -
Fe(II) Fe(III)
Southern Illinois University Carbondale OpenSIUC Publications Department of Microbiology 2006 Microbes Pumping Iron: Anaerobic Microbial Iron Oxidation and Reduction Karrie A. Weber University of California - Berkeley Laurie A. Achenbach Southern Illinois University Carbondale, [email protected] John D. Coates University of California - Berkeley Follow this and additional works at: https://opensiuc.lib.siu.edu/micro_pubs Published in Nature Reviews Microbiology 4, 752-764 (October 2006), http://dx.doi.org/10.1038/ nrmicro1490. This Article is brought to you for free and open access by the Department of Microbiology at OpenSIUC. It has been accepted for inclusion in Publications by an authorized administrator of OpenSIUC. For more information, please contact [email protected]. 1 Microbes Pumping Iron: 2 Anaerobic Microbial Iron Oxidation and Reduction 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Karrie A. Weber1, Laurie A. Achenbach2, and John D. Coates1* 18 19 20 21 22 23 1Department of Plant and Microbial Biology 24 271 Koshland Hall 25 University of California, Berkeley 26 Berkeley, CA 94720 27 28 29 2Department of Microbiology 30 Southern Illinois University 31 Carbondale, IL 62901 32 33 34 35 Corresponding Author 36 Tel: (510) 643-8455 37 Fax: (510) 642-4995 38 e-mail: [email protected] 39 1 1 Abstract 2 Iron (Fe) has long been a recognized physiological requirement for life, yet its role for a 3 broad diversity of microorganisms persisting in water, soils, and sediments extends well-beyond 4 a nutritional necessity. Microorganisms from within both the domain Archaea and Bacteria are 5 capable of metabolically exploiting the favorable redox potential between the Fe(III)/Fe(II) 6 couple. -

Supplementary Material Impacts of Radiation on the Bacterial And
Supplementary Material Impacts of radiation on the bacterial and fungal microbiome of small mammals in the Chernobyl Exclusion Zone Rachael E. Antwis, Nicholas A. Beresford, Joseph A. Jackson, Ross Fawkes, Catherine L. Barnett, Elaine Potter, Lee Walker, Sergey Gaschak, Michael D. Wood Table S1 Sample sizes of host species used in the study, and associated sex, total absorbed dose rate, burn and site categories for these samples. Animals estimated to receive total absorbed dose rates of <4 µGy h-1 were assigned ‘low’, those with estimated dose rates of 4-42 µGy h-1 assigned ‘medium’, and those >42 µGy h-1 assigned to the ‘high’ category. All samples from 2017 were collected within the Red Forest but from areas that had experienced different degrees of damage from forest fires. Samples from 2018 were either collected within or outside the Red Forest. Sampling information Sex Total absorbed dose category Burn category Site category Year Sample Host species Total n Female Male Unidentified Low Medium High Burnt Burnt Unburnt Inside Red Outside type (regrowth) (minimal Forest Red regrowth) Forest 2017 Faeces Bank vole 22 5 16 1 0 0 22 3 1 18 All - 2017 Faeces Striped field mouse 29 10 18 1 0 7 22 15 0 14 All - 2017 Faeces Wood mouse 27 6 20 1 0 0 27 2 25 0 All - 2017 Faeces Yellow-necked mouse 58 28 29 1 0 7 51 19 17 22 All - 2018 Gut Bank vole 142 59 83 0 42 64 36 - - - 34 108 (caecum) RESULTS Differences in community composition between host species and sample types Wood mice and yellow-necked mice had a similar bacterial community composition to one another (Figure 1a), with the exception of the family Helicobacteraceae (Z = 2.982, p = 0.023; Figure 1c). -

Bacterial Diversity in the Intestine of Sea Cucumber Stichopus Japonicus
Bacterial diversity in the intestine of sea cucumber Stichopus japonicus Item Type article Authors Gao, M.L.; Hou, H.M.; Zhang, G.L.; Liu, Y.; Sun, L.M. Download date 29/09/2021 15:37:50 Link to Item http://hdl.handle.net/1834/37808 Iranian Journal of Fisheries Sciences 16(1)318-325 2017 Bacterial diversity in the intestine of sea cucumber Stichopus japonicus * Gao M.L.; Hou H.M. ; Zhang G.L.; Liu Y.; Sun L.M. Received: May 2015 Accepted: August 2016 Abstract The intestinal bacterial diversity of Stichopus japonicus was investigated using 16S ribosomal RNA gene (rDNA) clone library and Polymerase Chain Reaction/Denaturing Gradient Gel Electrophoresis (PCR-DGGE). The clone library yielded a total of 188 clones, and these were sequenced and classified into 106 operational taxonomic units (OTUs) with sequence similarity ranging from 88 to 100%. The coverage of the library was 77.4%, with approximately 88.7% of the sequences affiliated to Proteobacteria. Gammaproteobacteria and Vibrio sp. were the predominant groups in the intestine of S. japonicus. Some bacteria such as Legionella sp., Brachybacterium sp., Streptomyces sp., Propionigenium sp. and Psychrobacter sp were first identified in the intestine of sea cucumber. Keywords: Intestinal bacterial diversity, 16S rDNA, PCR-DGGE, Sequencing, Stichopus japonicus School of Food Science and Technology- Dalian Polytechnic University, Dalian 116034, P. R. China * Corresponding author's Email:[email protected] 319 Gao et al., Bacterial diversity in the intestine of sea cucumber Stichopus japonicas Introduction In this paper, 16S rDNA clone Sea cucumber Stichopus japonicus is library analysis approaches and one of the most important holothurian PCR-DGGE fingerprinting of the 16S species in coastal fisheries. -
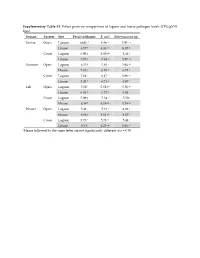
Supplementary Table S1. Fisher Pairwise Comparisons of Lagoon and House Pathogen Levels (CFU/Gvss Log10) Season System Site Fecal Coliforms E
Supplementary Table S1. Fisher pairwise comparisons of lagoon and house pathogen levels (CFU/gVSS log10) Season System Site Fecal coliforms E. coli Enterococcus sp. Spring Open Lagoon 6.00 g1 5.46 hi 5.91 cd House 6.57 e 6.33 cd 6.35 b Cover Lagoon 6.00 g 5.60 gh 5.44 f House 7.73 a 7.63 a 5.87 cd Summer Open Lagoon 6.35 f 5.83 f 5.94 cd House 7.83 a 6.10 e 6.73 a Cover Lagoon 7.04 c 6.47 c 5.86 cd House 7.42 b 6.71 b 6.07 c Fall Open Lagoon 5.58 i 5.58 gh 5.56 ef House 6.81 d 6.72 b 6.01 c Cover Lagoon 5.99 g 5.34 i 5.50 f House 6.38 f 6.19 de 5.74 de Winter Open Lagoon 5.41 j 5.13 j 4.88 g House 6.06 g 5.61 gh 6.67 a Cover Lagoon 5.73 h 5.73 fg 5.44 f House 6.34 f 6.25 de 5.86 cd 1Means followed by the same letter are not significantly different at p = 0.05 Supplementary Table S2. Relative abundances of OTUs identified using universal bacterial primer set, presented as relative abundances (%) Only bacterial families are counted in Figure 5 and discussion involving bacterial family identification. # Identified in # Identified from all 8 All 16 All #OTU Table SpOLRA SuOLRA FOLRA WOLRA SpCLRA SuCLRA FCLRA WCLRA SpOHRA SuOHRA FOHRA WOHRA SpCHRA SuCHRA FCHRA WCHRA all samples lagoon samples Samples Lagoons Notes Key Patulibacteraceae 0 9.93739E‐05* 0.000134 0.000116 0 0 0 0 0 0 0.000452 0 0 0 0 0 4 3 Sp = Spring O = Open Ruaniaceae 0 0 0 0 0 0 0 0 0 0 0 0 0 0.000221577 0 7.73E‐05 2 0 Su = Summer C = Covered Thermoactinomycetaceae 0 0 0.000267 0.000116 0 0.00037092 0.000529 0.000526 0 0.000342922 0.000271 0.000197 0 0 0 0 8 5 F = Fall L = Lagoon Beutenbergiaceae 0.000226334 0.000331246 0.000579 0.001663 0.000620176 0 0.000106 0.000807 0.002320743 0.000571537 0.000723 0.000591 0.000169731 0 0.000606 0 13 7 W = Winter H = House Thermoanaerobacterales Family III. -

Molecular Analyses of Changes Induced in the Microbial Populations of Murine Colon After As(III) Exposure Rishu Dheer
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Summer 2011 Molecular Analyses of Changes Induced in the Microbial Populations of Murine Colon After As(III) Exposure Rishu Dheer Follow this and additional works at: https://dsc.duq.edu/etd Recommended Citation Dheer, R. (2011). Molecular Analyses of Changes Induced in the Microbial Populations of Murine Colon After As(III) Exposure (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/484 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. For more information, please contact [email protected]. MOLECULAR ANALYSES OF CHANGES INDUCED IN THE MICROBIAL POPULATIONS OF MURINE COLON AFTER As (III) EXPOSURE A Dissertation Submitted to Bayer School of Natural and Environmental Sciences Duquesne University In partial fulfillment of the requirements for the degree of Doctor of Philosophy By Rishu Dheer August, 2011 Copyright by Rishu Dheer 2011 MOLECULAR ANALYSES OF CHANGES INDUCED IN THE MICROBIAL POPULATIONS OF MURINE COLON AFTER As (III) EXPOSURE By Rishu Dheer Approved June 14, 2011 ________________________________ ________________________________ Dr. John F. Stolz Dr. Aaron Barchowsky Director, Center of Environmental Associate Professor of Biology Research and Education (Committee Member) Professor of Biology (Committee Chair) ________________________________ -

Oral Sub-Chronic Ochratoxin a Exposure Induces Gut Microbiota Alterations in Mice
toxins Article Oral Sub-Chronic Ochratoxin a Exposure Induces Gut Microbiota Alterations in Mice María Izco 1 , Ariane Vettorazzi 2,3 , Maria de Toro 4, Yolanda Sáenz 5 and Lydia Alvarez-Erviti 1,* 1 Laboratory of Molecular Neurobiology, Center for Biomedical Research of La Rioja (CIBIR), 26006 Logroño, Spain; [email protected] 2 MITOX Group, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Universidad de Navarra, 31008 Pamplona, Spain; [email protected] 3 IdiSNA, Navarra Institute for Health Research, 31008 Pamplona, Spain 4 Center for Biomedical Research of La Rioja (CIBIR), Genomics and Bioinformatics Core Facility, 26006 Logroño, Spain; [email protected] 5 Molecular Microbiology Area, Center for Biomedical Research of La Rioja (CIBIR), 26006 Logroño, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-941-278-875 Abstract: Gut microbiota plays crucial roles in maintaining host health. External factors, such as diet, medicines, and environmental toxins, influence the composition of gut microbiota. Ochratoxin A (OTA) is one of the most prevalent and relevant mycotoxins and is a highly abundant food and animal feed contaminant. In the present study, we aimed to investigate OTA gut microbiome toxicity in mice sub-chronically exposed to low doses of OTA (0.21, 0.5, and 1.5 mg/kg body weight) by daily oral gavage for 28 days. Fecal microbiota from control and OTA-treated mice was analyzed using 16S ribosomal RNA (rRNA) gene sequencing followed by metagenomics. OTA exposure caused marked changes in gut microbial community structure, including the decrease in the diversity of fecal microbiota and the relative abundance of Firmicutes, as well as the increase in the relative abundance of Bacteroidetes at the phylum level. -

Taxonomic Hierarchy of the Phylum Proteobacteria and Korean Indigenous Novel Proteobacteria Species
Journal of Species Research 8(2):197-214, 2019 Taxonomic hierarchy of the phylum Proteobacteria and Korean indigenous novel Proteobacteria species Chi Nam Seong1,*, Mi Sun Kim1, Joo Won Kang1 and Hee-Moon Park2 1Department of Biology, College of Life Science and Natural Resources, Sunchon National University, Suncheon 57922, Republic of Korea 2Department of Microbiology & Molecular Biology, College of Bioscience and Biotechnology, Chungnam National University, Daejeon 34134, Republic of Korea *Correspondent: [email protected] The taxonomic hierarchy of the phylum Proteobacteria was assessed, after which the isolation and classification state of Proteobacteria species with valid names for Korean indigenous isolates were studied. The hierarchical taxonomic system of the phylum Proteobacteria began in 1809 when the genus Polyangium was first reported and has been generally adopted from 2001 based on the road map of Bergey’s Manual of Systematic Bacteriology. Until February 2018, the phylum Proteobacteria consisted of eight classes, 44 orders, 120 families, and more than 1,000 genera. Proteobacteria species isolated from various environments in Korea have been reported since 1999, and 644 species have been approved as of February 2018. In this study, all novel Proteobacteria species from Korean environments were affiliated with four classes, 25 orders, 65 families, and 261 genera. A total of 304 species belonged to the class Alphaproteobacteria, 257 species to the class Gammaproteobacteria, 82 species to the class Betaproteobacteria, and one species to the class Epsilonproteobacteria. The predominant orders were Rhodobacterales, Sphingomonadales, Burkholderiales, Lysobacterales and Alteromonadales. The most diverse and greatest number of novel Proteobacteria species were isolated from marine environments. Proteobacteria species were isolated from the whole territory of Korea, with especially large numbers from the regions of Chungnam/Daejeon, Gyeonggi/Seoul/Incheon, and Jeonnam/Gwangju. -

Gut Microbiota Analysis UC Davis MMPC - Microbiome & Host Response Core
D4006- Gut Microbiota Analysis UC Davis MMPC - Microbiome & Host Response Core Contents 1 Methods: 1 1.1 Sequencing . .1 1.2 Data processing . .1 2 Summary of Findings: 2 2.1 Sequencing analysis . .2 2.2 Microbial diversity . .2 2.2.1 Alpha Diversity . .2 2.2.2 Beta Diversity . 10 2.3 Data analysis using taxa abundance data . 13 2.3.1 Stacked bar graphs of Taxa abundances at each level . 14 A Appendix 1 (Taxa Abundance Tables) 30 B Appendix 2 (Taxa removed from filtered datasets at each level) 37 References: 43 Core Contacts: Helen E. Raybould, Ph.D., Core Leader ([email protected]) Trina A. Knotts, Ph.D., Core Co-Leader ([email protected]) Michael L. Goodson, Ph.D., Core Scientist ([email protected]) Client(s): Kent Lloyd, DVM PhD ;MMRRC; UC Davis Project #: MBP-2079 MMRRC strain ID: MMRRC_043568 Animal Information: The strain was donated to the MMRRC by Michael Mitchell at Inserm. Fecal samples were obtained from animals housed under the care of Arthur Arnold at UCLA. 1 Methods: Brief Project Description: MMRRC strains are often contributed to the MMRRC to fulfill the resource sharing aspects of NIH grants. Since transporting mice to another facilty often causes a microbiota shift, having a record of the original fecal microbiota from the donor institution where the original phenotyping or testing was performed may prove helpful if a phenotype is lost after transfer. Several MMRRC mouse lines were selected for fecal microbiota profiling of the microbiota. Table 1: Animal-Strain Information X.SampleID TreatmentGroup Animal_ID Genotype Line Sex MMRRC.043568.M93054 MMRRC.043568_Tg.plus_M M93054 Tg.plus MMRRC.043568 M MMRRC.043568.M06285 MMRRC.043568_Tg.plus_M M06285 Tg.plus MMRRC.043568 M 1 1.1 Sequencing Frozen fecal or regional gut samples were shipped on dry ice to UC Davis MMPC and Host Microbe Systems Biology Core.