Dddy Is a Bacterial Dimethylsulfoniopropionate Lyase Representing a New Cupin Enzyme Superfamily with Unknown Primary Function
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Gut Microbiome of the Sea Urchin, Lytechinus Variegatus, from Its Natural Habitat Demonstrates Selective Attributes of Micro
FEMS Microbiology Ecology, 92, 2016, fiw146 doi: 10.1093/femsec/fiw146 Advance Access Publication Date: 1 July 2016 Research Article RESEARCH ARTICLE The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles Joseph A. Hakim1,†, Hyunmin Koo1,†, Ranjit Kumar2, Elliot J. Lefkowitz2,3, Casey D. Morrow4, Mickie L. Powell1, Stephen A. Watts1,∗ and Asim K. Bej1,∗ 1Department of Biology, University of Alabama at Birmingham, 1300 University Blvd, Birmingham, AL 35294, USA, 2Center for Clinical and Translational Sciences, University of Alabama at Birmingham, Birmingham, AL 35294, USA, 3Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL 35294, USA and 4Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, 1918 University Blvd., Birmingham, AL 35294, USA ∗Corresponding authors: Department of Biology, University of Alabama at Birmingham, 1300 University Blvd, CH464, Birmingham, AL 35294-1170, USA. Tel: +1-(205)-934-8308; Fax: +1-(205)-975-6097; E-mail: [email protected]; [email protected] †These authors contributed equally to this work. One sentence summary: This study describes the distribution of microbiota, and their predicted functional attributes, in the gut ecosystem of sea urchin, Lytechinus variegatus, from its natural habitat of Gulf of Mexico. Editor: Julian Marchesi ABSTRACT In this paper, we describe the microbial composition and their predictive metabolic profile in the sea urchin Lytechinus variegatus gut ecosystem along with samples from its habitat by using NextGen amplicon sequencing and downstream bioinformatics analyses. The microbial communities of the gut tissue revealed a near-exclusive abundance of Campylobacteraceae, whereas the pharynx tissue consisted of Tenericutes, followed by Gamma-, Alpha- and Epsilonproteobacteria at approximately equal capacities. -

Thalassomonas Agarivorans Sp. Nov., a Marine Agarolytic Bacterium Isolated from Shallow Coastal Water of An-Ping Harbour, Taiwan
International Journal of Systematic and Evolutionary Microbiology (2006), 56, 1245–1250 DOI 10.1099/ijs.0.64130-0 Thalassomonas agarivorans sp. nov., a marine agarolytic bacterium isolated from shallow coastal water of An-Ping Harbour, Taiwan, and emended description of the genus Thalassomonas Wen Dar Jean,1 Wung Yang Shieh2 and Tung Yen Liu2 Correspondence 1Center for General Education, Leader University, No. 188, Sec. 5, An-Chung Rd, Tainan, Wung Yang Shieh Taiwan [email protected] 2Institute of Oceanography, National Taiwan University, PO Box 23-13, Taipei, Taiwan A marine agarolytic bacterium, designated strain TMA1T, was isolated from a seawater sample collected in a shallow-water region of An-Ping Harbour, Taiwan. It was non-fermentative and Gram-negative. Cells grown in broth cultures were straight or curved rods, non-motile and non-flagellated. The isolate required NaCl for growth and exhibited optimal growth at 25 6C and 3 % NaCl. It grew aerobically and was incapable of anaerobic growth by fermenting glucose or other carbohydrates. Predominant cellular fatty acids were C16 : 0 (17?5 %), C17 : 1v8c (12?8 %), C17 : 0 (11?1 %), C15 : 0 iso 2-OH/C16 : 1v7c (8?6 %) and C13 : 0 (7?3 %). The DNA G+C content was 41?0 mol%. Phylogenetic, phenotypic and chemotaxonomic data accumulated in this study revealed that the isolate could be classified in a novel species of the genus Thalassomonas in the family Colwelliaceae. The name Thalassomonas agarivorans sp. nov. is proposed for the novel species, with TMA1T (=BCRC 17492T=JCM 13379T) as the type strain. Alteromonas-like bacteria in the class Gammaproteobacteria however, they are not exclusively autochthonous in the comprise a large group of marine, heterotrophic, polar- marine environment, since some reports have shown that flagellated, Gram-negative rods that are mainly non- they also occur in freshwater, sewage and soil (Agbo & Moss, fermentative aerobes. -

Bacterial Diversity in the Intestine of Sea Cucumber Stichopus Japonicus
Bacterial diversity in the intestine of sea cucumber Stichopus japonicus Item Type article Authors Gao, M.L.; Hou, H.M.; Zhang, G.L.; Liu, Y.; Sun, L.M. Download date 29/09/2021 15:37:50 Link to Item http://hdl.handle.net/1834/37808 Iranian Journal of Fisheries Sciences 16(1)318-325 2017 Bacterial diversity in the intestine of sea cucumber Stichopus japonicus * Gao M.L.; Hou H.M. ; Zhang G.L.; Liu Y.; Sun L.M. Received: May 2015 Accepted: August 2016 Abstract The intestinal bacterial diversity of Stichopus japonicus was investigated using 16S ribosomal RNA gene (rDNA) clone library and Polymerase Chain Reaction/Denaturing Gradient Gel Electrophoresis (PCR-DGGE). The clone library yielded a total of 188 clones, and these were sequenced and classified into 106 operational taxonomic units (OTUs) with sequence similarity ranging from 88 to 100%. The coverage of the library was 77.4%, with approximately 88.7% of the sequences affiliated to Proteobacteria. Gammaproteobacteria and Vibrio sp. were the predominant groups in the intestine of S. japonicus. Some bacteria such as Legionella sp., Brachybacterium sp., Streptomyces sp., Propionigenium sp. and Psychrobacter sp were first identified in the intestine of sea cucumber. Keywords: Intestinal bacterial diversity, 16S rDNA, PCR-DGGE, Sequencing, Stichopus japonicus School of Food Science and Technology- Dalian Polytechnic University, Dalian 116034, P. R. China * Corresponding author's Email:[email protected] 319 Gao et al., Bacterial diversity in the intestine of sea cucumber Stichopus japonicas Introduction In this paper, 16S rDNA clone Sea cucumber Stichopus japonicus is library analysis approaches and one of the most important holothurian PCR-DGGE fingerprinting of the 16S species in coastal fisheries. -

Pan-Genome Analyses of 24 Shewanella Strains Re-Emphasize
Zhong et al. Biotechnol Biofuels (2018) 11:193 https://doi.org/10.1186/s13068-018-1201-1 Biotechnology for Biofuels RESEARCH Open Access Pan‑genome analyses of 24 Shewanella strains re‑emphasize the diversifcation of their functions yet evolutionary dynamics of metal‑reducing pathway Chaofang Zhong, Maozhen Han, Shaojun Yu, Pengshuo Yang, Hongjun Li and Kang Ning* Abstract Background: Shewanella strains are important dissimilatory metal-reducing bacteria which are widely distributed in diverse habitats. Despite eforts to genomically characterize Shewanella, knowledge of the molecular components, functional information and evolutionary patterns remain lacking, especially for their compatibility in the metal- reducing pathway. The increasing number of genome sequences of Shewanella strains ofers a basis for pan-genome studies. Results: A comparative pan-genome analysis was conducted to study genomic diversity and evolutionary relation- ships among 24 Shewanella strains. Results revealed an open pan-genome of 13,406 non-redundant genes and a core-genome of 1878 non-redundant genes. Selective pressure acted on the invariant members of core genome, in which purifying selection drove evolution in the housekeeping mechanisms. Shewanella strains exhibited extensive genome variability, with high levels of gene gain and loss during the evolution, which afected variable gene sets and facilitated the rapid evolution. Additionally, genes related to metal reduction were diversely distributed in Shewanella strains and evolved under purifying selection, which highlighted the basic conserved functionality and specifcity of respiratory systems. Conclusions: The diversity of genes present in the accessory and specifc genomes of Shewanella strains indicates that each strain uses diferent strategies to adapt to diverse environments. -

Taxonomic Hierarchy of the Phylum Proteobacteria and Korean Indigenous Novel Proteobacteria Species
Journal of Species Research 8(2):197-214, 2019 Taxonomic hierarchy of the phylum Proteobacteria and Korean indigenous novel Proteobacteria species Chi Nam Seong1,*, Mi Sun Kim1, Joo Won Kang1 and Hee-Moon Park2 1Department of Biology, College of Life Science and Natural Resources, Sunchon National University, Suncheon 57922, Republic of Korea 2Department of Microbiology & Molecular Biology, College of Bioscience and Biotechnology, Chungnam National University, Daejeon 34134, Republic of Korea *Correspondent: [email protected] The taxonomic hierarchy of the phylum Proteobacteria was assessed, after which the isolation and classification state of Proteobacteria species with valid names for Korean indigenous isolates were studied. The hierarchical taxonomic system of the phylum Proteobacteria began in 1809 when the genus Polyangium was first reported and has been generally adopted from 2001 based on the road map of Bergey’s Manual of Systematic Bacteriology. Until February 2018, the phylum Proteobacteria consisted of eight classes, 44 orders, 120 families, and more than 1,000 genera. Proteobacteria species isolated from various environments in Korea have been reported since 1999, and 644 species have been approved as of February 2018. In this study, all novel Proteobacteria species from Korean environments were affiliated with four classes, 25 orders, 65 families, and 261 genera. A total of 304 species belonged to the class Alphaproteobacteria, 257 species to the class Gammaproteobacteria, 82 species to the class Betaproteobacteria, and one species to the class Epsilonproteobacteria. The predominant orders were Rhodobacterales, Sphingomonadales, Burkholderiales, Lysobacterales and Alteromonadales. The most diverse and greatest number of novel Proteobacteria species were isolated from marine environments. Proteobacteria species were isolated from the whole territory of Korea, with especially large numbers from the regions of Chungnam/Daejeon, Gyeonggi/Seoul/Incheon, and Jeonnam/Gwangju. -

Ferrimonas Balearica Type Strain (PAT)
Lawrence Berkeley National Laboratory Recent Work Title Complete genome sequence of Ferrimonas balearica type strain (PAT). Permalink https://escholarship.org/uc/item/9hf3t0j0 Journal Standards in genomic sciences, 3(2) ISSN 1944-3277 Authors Nolan, Matt Sikorski, Johannes Davenport, Karen et al. Publication Date 2010 DOI 10.4056/sigs.1161239 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Standards in Genomic Sciences (2010) 3:174-182 DOI:10.4056/sigs.1161239 Complete genome sequence of Ferrimonas balearica type strain (PATT) Matt Nolan1, Johannes Sikorski2, Karen Davenport1,3, Susan Lucas1, Tijana Glavina Del Rio1, Hope Tice1, Jan-Fang Cheng1, Lynne Goodwin1,3, Sam Pitluck1, Konstantinos Liolios1, Natalia Ivanova1, Konstantinos Mavromatis1, Galina Ovchinnikova1, Amrita Pati1, Amy Chen4, Krishna Palaniappan4, Miriam Land1,5, Loren Hauser1,5, Yun-Juan Chang1,5, Cynthia D. Jef- fries1,5, Roxanne Tapia1,3, Thomas Brettin1,3, John C. Detter1,3, Cliff Han1,3, Montri Yasa- wong6, Manfred Rohde6, Brian J Tindall2, Markus Göker2, Tanja Woyke1, James Bristow1, Jo- nathan A. Eisen1,7, Victor Markowitz4, Philip Hugenholtz1, Nikos C. Kyrpides1, Hans-Peter Klenk2*, and Alla Lapidus1 1 DOE Joint Genome Institute, Walnut Creek, California, USA 2 DSMZ - German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany 3 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA 4 Biological Data Management and Technology Center, Lawrence Berkeley National Labora- tory, Berkeley, California, USA 5 Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA 6 HZI – Helmholtz Centre for Infection Research, Braunschweig, Germany 7 University of California Davis Genome Center, Davis, California, USA *Corresponding author: Hans-Peter Klenk Keywords: chemoorganotroph, iron(III)-reducing bacterium, facultatively anaerobic, Ferrimo- nadaceae, Gammaproteobacteria, GEBA Ferrimonas balearica Rossello-Mora et al. -
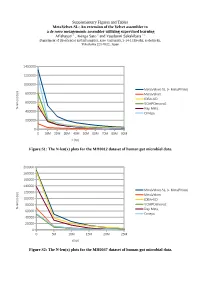
Supplementary Figures and Tables Metavelvet-SL
Supplementary Figures and Tables MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning Afiahayati 1 , Kengo Sato 1 and Yasubumi Sakakibara 1∗ Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan 1400000 1200000 1000000 MetaVelvet-SL (+ MetaPhlAn) ) p 800000 b MetaVelvet ( ) x ( IDBA-UD n 600000 e SOAPDenovo2 l - N Ray Meta 400000 Omega 200000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S1: The N-len(x) plots for the MH0012 dataset of human gut microbial data. 200000 180000 160000 140000 MetaVelvet-SL (+ MetaPhlAn) ) 120000 p b MetaVelvet ( ) 100000 x IDBA-UD ( n e l 80000 SOAPDenovo2 - N Ray Meta 60000 Omega 40000 20000 0 0 5M 10M 15M 20M 25M x(bp) Figure S2: The N-len(x) plots for the MH0047 dataset of human gut microbial data. 600000 500000 400000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 300000 x IDBA-UD ( n e l SOAPDenovo2 - N 200000 Ray Meta Omega 100000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S3: The N-len(x) plots for the SRS017227 dataset of human gut microbial data. 800000 700000 600000 500000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 400000 x IDBA-UD ( n e l SOAPDenovo2 - 300000 N Ray Meta 200000 Omega 100000 0 0 5M 10M 15M 20M 25M 30M 35M 40M 45M x (bp) Figure S4: The N-len(x) plots for the SRS018661 dataset of human gut microbial data. Formula to identify unique nodes in Velvet (Zerbino and Birney, 2008) ̄x2 ρ2− log2 2 F ( ̄x ,n,ρ )= +n 2 2 where ̄x = the coverage of node ρ = expected coverage of subgraph n = the length of node A node is “unique”, if its F > 5 . -

10 the Family Colwelliaceae John P
10 The Family Colwelliaceae John P. Bowman Food Safety Centre, Tasmanian Institute of Agriculture, University of Tasmania, Hobart, TAS, Australia Taxonomy, Historical, and Current Short Description Taxonomy, Historical, and Current Short of the Family Colwelliaceae Ivanova Description of the Family Colwelliaceae VP et al. 2004, 1773VP .........................................179 Ivanova et al. 2004, 1773 Molecular Analyses . ..................................180 Col.well.i0a.ce.ae. N.L. fem. n. Colwellia, type genus of the fam- ily; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Phenotypic Properties . ..................................181 Colwelliaceae, the Colwellia family. The family Colwelliaceae was first described by Ivanova and Genus Colwellia Deming et al. 1988, 328AL ...............181 colleagues (2004) as part of an effort to create taxonomic har- mony within a large clade of almost exclusively marine bacteria Genus Thalassomonas Macia´n et al. 2001, 1283 . 187 located within class Gammaproteobacteria. This clade now rep- resents the order Alteromonadales (Bowman and McMeekin Enrichment, Isolation, and Maintenance Procedures . 187 2005) and consists of at least 22 genera as of late 2012. In addition to the genera Colwellia and Thalassomonas, Genome-Based and Genetic Studies . .....................191 the other members of the order include Aesturaiibacter, Agarivorans, Algicola, Aliagarivorans, Alkalimonas, Ecology . ...............................................192 Alishewanella, Alteromonas, Bowmanella, Catenovulum, -

Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina Cyanorosea
microorganisms Article Not That Close to Mommy: Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina cyanorosea Bruno F. R. Oliveira 1,2 , Isabelle R. Lopes 1, Anna L. B. Canellas 1 , Guilherme Muricy 3, Alan D. W. Dobson 2,4 and Marinella S. Laport 1,* 1 Laboratório de Bacteriologia Molecular e Marinha, Instituto de Microbiologia Paulo de Góes, Universidade Federal do Rio de Janeiro, Rio de Janeiro 21941902, Brazil; [email protected] (B.F.R.O.); [email protected] (I.R.L.); [email protected] (A.L.B.C.) 2 School of Microbiology, University College Cork, T12 Y960 Cork, Ireland; [email protected] 3 Laboratório de Biologia de Porifera, Museu Nacional, Universidade Federal do Rio de Janeiro, Rio de Janeiro 20940040, Brazil; [email protected] 4 Environmental Research Institute, University College Cork, T23 XE10 Cork, Ireland * Correspondence: [email protected] Received: 18 September 2020; Accepted: 25 November 2020; Published: 12 December 2020 Abstract: Marine sponges are excellent examples of invertebrate–microbe symbioses. In this holobiont, the partnership has elegantly evolved by either transmitting key microbial associates through the host germline and/or capturing microorganisms from the surrounding seawater. We report here on the prokaryotic microbiota during different developmental stages of Plakina cyanorosea and their surrounding environmental samples by a 16S rRNA metabarcoding approach. In comparison with their source adults, larvae housed slightly richer and more diverse microbial communities, which are structurally more related to the environmental microbiota. In addition to the thaumarchaeal Nitrosopumilus, parental sponges were broadly dominated by Alpha- and Gamma-proteobacteria, while the offspring were particularly enriched in the Vibrionales, Alteromonodales, Enterobacterales orders and the Clostridia and Bacteroidia classes. -

Trophic Niches Reflect Compositional Differences in Microbiota Among Caribbean Sea Urchins
Trophic niches reflect compositional differences in microbiota among Caribbean sea urchins Ruber Rodríguez-Barreras1, Eduardo L. Tosado-Rodríguez2 and Filipa Godoy-Vitorino2 1 Department of Biology, University of Puerto Rico at Bayamón, Bayamón, Puerto Rico, USA 2 Microbiology and Medical Zoology, School of Medicine, University of Puerto Rico, School of Medicine, San Juan, Puerto Rico, USA ABSTRACT Sea urchins play a critical role in marine ecosystems, as they actively participate in maintaining the balance between coral and algae. We performed the first in-depth survey of the microbiota associated with four free-living populations of Caribbean sea urchins: Lytechinus variegatus, Echinometra lucunter, Tripneustes ventricosus, and Diadema antillarum. We compared the influence of the collection site, echinoid species and trophic niche to the composition of the microbiota. This dataset provides a comprehensive overview to date, of the bacterial communities and their ecological relevance associated with sea urchins in their natural environments. A total of sixty- samples, including surrounding reef water and seagrass leaves underwent 16S rRNA gene sequencing (V4 region) and high-quality reads were analyzed with standard bioinformatic approaches. While water and seagrass were dominated by Cyanobacteria such as Prochlorococcus and Rivularia respectively, echinoid gut samples had dominant Bacteroidetes, Proteobacteria and Fusobacteria. Propionigenium was dominant across all species' guts, revealing a host-associated composition likely responsive to the digestive process of the animals. Beta-diversity analyses showed significant differences Submitted 24 March 2021 in community composition among the three collection sites, animal species, and Accepted 7 August 2021 Published 31 August 2021 trophic niches. Alpha diversity was significantly higher among L. -

Isolation and Molecular Characterization of Microbial Population from the Fish
! Journal!of!Materials!and!! J. Mater. Environ. Sci., 2021, Volume 12, Issue 04, Page 573-583 Environmental!Science! ISSN!:!2028;2508! http://www.jmaterenvironsci.com CODEN!:!JMESCN! ! Copyright*©*2021,* University*of*Mohammed*Premier****** Oujda*Morocco* ! Isolation and molecular characterization of microbial population from the Fish ‘Tilapia’ collected from Vembanad Lake, Kerala, India! Hari Prabha1, Kallu Nataraj1, B.R. Rajesh2, R. Pratap Chandran2* 1S.N.G.M. Arts and Science College, Thuravoor, Cherthala, Kerala, India 2Department of Biotechnology and Research, K. V. M. College of Science and Technology, Kokkothamangalam P. O., Cherthala - 688527, Alappuzha District, Kerala State, India. ! Received 20 Feb 2021, Abstract Revised 13 April 2021, Water bodies provide a large! amount of easily accessible fresh water which stabilizes Accepted 15 April 2021 any population. Kuttanchaal is a beautiful island surrounded by Vembanad Lake, in Thycattusserry, Cherthala,! Alappuzha, Kerala, India. The major livelihood of the Keywords people living in this island is fishing and coir industry. For the study of the fish, swabs ! Fish from the face, gut and gills! were taken directly and analysed for microbial content. The ! Ferrimonas balearica, water sample was also subjected to physicochemical analysis. The temperature, ! Salmonella salinity and specific gravity of water sample was 32.7℃, 4-7 ppt and 1.004-1.007 ! pathogenic bacteria kg/m3 respectively. Bacteriological! analysis of water and fish samples were done using ! Vembanad Lake Thiosulphate Citrate Bile salt Sucrose (TCBS), Eosin Methylene Blue (EMB) and ! Bismuth Sulphite (BS) agar and the colonies were studied and antibiotic sensitivity ! assay was also done. Vibrio! cholerae and Ferrimonas balearica showed more [email protected] susceptibility towards Norfloxacin and least towards Furazolidone. -

Supplementary Materials
Supplementary Materials Supporting methods Sequence comparison to DNA isolation kit blank and drilling fluid (For Costa Rica sediment samples) Because DNA concentrations were very low in many of the sediment samples, and PCR tests indicated that in a few of the samples, if present at all, DNA may not be in high enough amounts to overcome the “background” DNA from the DNA extraction kits, a representative DNA extraction kit blank was sequenced along with all other samples. To remove any signal from the extraction kit in all samples, as well as to remove any samples whose genuine DNA was not in high enough abundance to overcome the extraction kit background, sequence results from the SILVA pipeline were processed initially as follows: 1. Classification of reads was examined at the “fully expanded” taxonomic depth from the SILVA pipeline output, and all lineages present in the extraction blank in any amount were flagged. 2. To account for sequencing error in classification, further lineages were added to the flagged ones by going up in taxonomic level to “order” and flagging every sequence identified as being from the same order as any sequence present in the extraction blank. There were a few cases where the taxonomy of sequences in the extraction blank did not go down as far as the level of "order", and for those, the most specific level identified above order was used to assess any further matches. For example, if the sequence was classified down to "class," then any remaining sequences in that class would also be removed. Those cases