Gut Microbiota Analysis UC Davis MMPC - Microbiome & Host Response Core
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

26Sor Em2rray /Mmucd
D4006- Gut Microbiota Analysis UC Davis MMPC - Microbiome & Host Response Core Contents 1 Methods: 1 1.1 Sequencing . .1 1.2 Data processing . .1 2 Summary of Findings: 2 2.1 Sequencing analysis . .2 2.2 Microbial diversity . .2 2.2.1 Alpha Diversity . .2 2.2.2 Beta Diversity . 10 2.3 Data analysis using taxa abundance data . 13 2.3.1 Stacked bar graphs of Taxa abundances at each level . 14 A Appendix 1 (Taxa Abundance Tables) 30 B Appendix 2 (Taxa removed from filtered datasets at each level) 37 References: 43 Core Contacts: Helen E. Raybould, Ph.D., Core Leader ([email protected]) Trina A. Knotts, Ph.D., Core Co-Leader ([email protected]) Michael L. Goodson, Ph.D., Core Scientist ([email protected]) Client(s): Kent Lloyd, DVM PhD ;MMRRC; UC Davis Project #: MBP-2079 MMRRC strain ID: MMRRC_043514 Animal Information: The strain was donated to the MMRRC by Russell Ray at Baylor College of Medicine. Fecal samples were obtained from animals housed under the care of Russell Ray at Baylor College of Medicine. 1 Methods: Brief Project Description: MMRRC strains are often contributed to the MMRRC to fulfill the resource sharing aspects of NIH grants. Since transporting mice to another facilty often causes a microbiota shift, having a record of the original fecal microbiota from the donor institution where the original phenotyping or testing was performed may prove helpful if a phenotype is lost after transfer. Several MMRRC mouse lines were selected for fecal microbiota profiling of the microbiota. Table 1: Animal-Strain Information X.SampleID TreatmentGroup Animal_ID Genotype Line Sex MMRRC.043514.Mx054 MMRRC.043514_Het_M Mx054 Het MMRRC.043514 M MMRRC.043514.Mx415 MMRRC.043514_Het_M Mx415 Het MMRRC.043514 M 1 1.1 Sequencing Frozen fecal or regional gut samples were shipped on dry ice to UC Davis MMPC and Host Microbe Systems Biology Core. -
Fe(II) Fe(III)
Southern Illinois University Carbondale OpenSIUC Publications Department of Microbiology 2006 Microbes Pumping Iron: Anaerobic Microbial Iron Oxidation and Reduction Karrie A. Weber University of California - Berkeley Laurie A. Achenbach Southern Illinois University Carbondale, [email protected] John D. Coates University of California - Berkeley Follow this and additional works at: https://opensiuc.lib.siu.edu/micro_pubs Published in Nature Reviews Microbiology 4, 752-764 (October 2006), http://dx.doi.org/10.1038/ nrmicro1490. This Article is brought to you for free and open access by the Department of Microbiology at OpenSIUC. It has been accepted for inclusion in Publications by an authorized administrator of OpenSIUC. For more information, please contact [email protected]. 1 Microbes Pumping Iron: 2 Anaerobic Microbial Iron Oxidation and Reduction 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Karrie A. Weber1, Laurie A. Achenbach2, and John D. Coates1* 18 19 20 21 22 23 1Department of Plant and Microbial Biology 24 271 Koshland Hall 25 University of California, Berkeley 26 Berkeley, CA 94720 27 28 29 2Department of Microbiology 30 Southern Illinois University 31 Carbondale, IL 62901 32 33 34 35 Corresponding Author 36 Tel: (510) 643-8455 37 Fax: (510) 642-4995 38 e-mail: [email protected] 39 1 1 Abstract 2 Iron (Fe) has long been a recognized physiological requirement for life, yet its role for a 3 broad diversity of microorganisms persisting in water, soils, and sediments extends well-beyond 4 a nutritional necessity. Microorganisms from within both the domain Archaea and Bacteria are 5 capable of metabolically exploiting the favorable redox potential between the Fe(III)/Fe(II) 6 couple. -

Supplementary Material Impacts of Radiation on the Bacterial And
Supplementary Material Impacts of radiation on the bacterial and fungal microbiome of small mammals in the Chernobyl Exclusion Zone Rachael E. Antwis, Nicholas A. Beresford, Joseph A. Jackson, Ross Fawkes, Catherine L. Barnett, Elaine Potter, Lee Walker, Sergey Gaschak, Michael D. Wood Table S1 Sample sizes of host species used in the study, and associated sex, total absorbed dose rate, burn and site categories for these samples. Animals estimated to receive total absorbed dose rates of <4 µGy h-1 were assigned ‘low’, those with estimated dose rates of 4-42 µGy h-1 assigned ‘medium’, and those >42 µGy h-1 assigned to the ‘high’ category. All samples from 2017 were collected within the Red Forest but from areas that had experienced different degrees of damage from forest fires. Samples from 2018 were either collected within or outside the Red Forest. Sampling information Sex Total absorbed dose category Burn category Site category Year Sample Host species Total n Female Male Unidentified Low Medium High Burnt Burnt Unburnt Inside Red Outside type (regrowth) (minimal Forest Red regrowth) Forest 2017 Faeces Bank vole 22 5 16 1 0 0 22 3 1 18 All - 2017 Faeces Striped field mouse 29 10 18 1 0 7 22 15 0 14 All - 2017 Faeces Wood mouse 27 6 20 1 0 0 27 2 25 0 All - 2017 Faeces Yellow-necked mouse 58 28 29 1 0 7 51 19 17 22 All - 2018 Gut Bank vole 142 59 83 0 42 64 36 - - - 34 108 (caecum) RESULTS Differences in community composition between host species and sample types Wood mice and yellow-necked mice had a similar bacterial community composition to one another (Figure 1a), with the exception of the family Helicobacteraceae (Z = 2.982, p = 0.023; Figure 1c). -
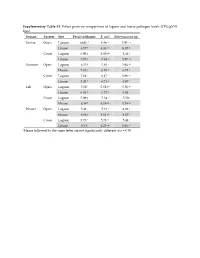
Supplementary Table S1. Fisher Pairwise Comparisons of Lagoon and House Pathogen Levels (CFU/Gvss Log10) Season System Site Fecal Coliforms E
Supplementary Table S1. Fisher pairwise comparisons of lagoon and house pathogen levels (CFU/gVSS log10) Season System Site Fecal coliforms E. coli Enterococcus sp. Spring Open Lagoon 6.00 g1 5.46 hi 5.91 cd House 6.57 e 6.33 cd 6.35 b Cover Lagoon 6.00 g 5.60 gh 5.44 f House 7.73 a 7.63 a 5.87 cd Summer Open Lagoon 6.35 f 5.83 f 5.94 cd House 7.83 a 6.10 e 6.73 a Cover Lagoon 7.04 c 6.47 c 5.86 cd House 7.42 b 6.71 b 6.07 c Fall Open Lagoon 5.58 i 5.58 gh 5.56 ef House 6.81 d 6.72 b 6.01 c Cover Lagoon 5.99 g 5.34 i 5.50 f House 6.38 f 6.19 de 5.74 de Winter Open Lagoon 5.41 j 5.13 j 4.88 g House 6.06 g 5.61 gh 6.67 a Cover Lagoon 5.73 h 5.73 fg 5.44 f House 6.34 f 6.25 de 5.86 cd 1Means followed by the same letter are not significantly different at p = 0.05 Supplementary Table S2. Relative abundances of OTUs identified using universal bacterial primer set, presented as relative abundances (%) Only bacterial families are counted in Figure 5 and discussion involving bacterial family identification. # Identified in # Identified from all 8 All 16 All #OTU Table SpOLRA SuOLRA FOLRA WOLRA SpCLRA SuCLRA FCLRA WCLRA SpOHRA SuOHRA FOHRA WOHRA SpCHRA SuCHRA FCHRA WCHRA all samples lagoon samples Samples Lagoons Notes Key Patulibacteraceae 0 9.93739E‐05* 0.000134 0.000116 0 0 0 0 0 0 0.000452 0 0 0 0 0 4 3 Sp = Spring O = Open Ruaniaceae 0 0 0 0 0 0 0 0 0 0 0 0 0 0.000221577 0 7.73E‐05 2 0 Su = Summer C = Covered Thermoactinomycetaceae 0 0 0.000267 0.000116 0 0.00037092 0.000529 0.000526 0 0.000342922 0.000271 0.000197 0 0 0 0 8 5 F = Fall L = Lagoon Beutenbergiaceae 0.000226334 0.000331246 0.000579 0.001663 0.000620176 0 0.000106 0.000807 0.002320743 0.000571537 0.000723 0.000591 0.000169731 0 0.000606 0 13 7 W = Winter H = House Thermoanaerobacterales Family III. -

Molecular Analyses of Changes Induced in the Microbial Populations of Murine Colon After As(III) Exposure Rishu Dheer
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Summer 2011 Molecular Analyses of Changes Induced in the Microbial Populations of Murine Colon After As(III) Exposure Rishu Dheer Follow this and additional works at: https://dsc.duq.edu/etd Recommended Citation Dheer, R. (2011). Molecular Analyses of Changes Induced in the Microbial Populations of Murine Colon After As(III) Exposure (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/484 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. For more information, please contact [email protected]. MOLECULAR ANALYSES OF CHANGES INDUCED IN THE MICROBIAL POPULATIONS OF MURINE COLON AFTER As (III) EXPOSURE A Dissertation Submitted to Bayer School of Natural and Environmental Sciences Duquesne University In partial fulfillment of the requirements for the degree of Doctor of Philosophy By Rishu Dheer August, 2011 Copyright by Rishu Dheer 2011 MOLECULAR ANALYSES OF CHANGES INDUCED IN THE MICROBIAL POPULATIONS OF MURINE COLON AFTER As (III) EXPOSURE By Rishu Dheer Approved June 14, 2011 ________________________________ ________________________________ Dr. John F. Stolz Dr. Aaron Barchowsky Director, Center of Environmental Associate Professor of Biology Research and Education (Committee Member) Professor of Biology (Committee Chair) ________________________________ -

Oral Sub-Chronic Ochratoxin a Exposure Induces Gut Microbiota Alterations in Mice
toxins Article Oral Sub-Chronic Ochratoxin a Exposure Induces Gut Microbiota Alterations in Mice María Izco 1 , Ariane Vettorazzi 2,3 , Maria de Toro 4, Yolanda Sáenz 5 and Lydia Alvarez-Erviti 1,* 1 Laboratory of Molecular Neurobiology, Center for Biomedical Research of La Rioja (CIBIR), 26006 Logroño, Spain; [email protected] 2 MITOX Group, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Universidad de Navarra, 31008 Pamplona, Spain; [email protected] 3 IdiSNA, Navarra Institute for Health Research, 31008 Pamplona, Spain 4 Center for Biomedical Research of La Rioja (CIBIR), Genomics and Bioinformatics Core Facility, 26006 Logroño, Spain; [email protected] 5 Molecular Microbiology Area, Center for Biomedical Research of La Rioja (CIBIR), 26006 Logroño, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-941-278-875 Abstract: Gut microbiota plays crucial roles in maintaining host health. External factors, such as diet, medicines, and environmental toxins, influence the composition of gut microbiota. Ochratoxin A (OTA) is one of the most prevalent and relevant mycotoxins and is a highly abundant food and animal feed contaminant. In the present study, we aimed to investigate OTA gut microbiome toxicity in mice sub-chronically exposed to low doses of OTA (0.21, 0.5, and 1.5 mg/kg body weight) by daily oral gavage for 28 days. Fecal microbiota from control and OTA-treated mice was analyzed using 16S ribosomal RNA (rRNA) gene sequencing followed by metagenomics. OTA exposure caused marked changes in gut microbial community structure, including the decrease in the diversity of fecal microbiota and the relative abundance of Firmicutes, as well as the increase in the relative abundance of Bacteroidetes at the phylum level. -

Pan-Genome Analyses of 24 Shewanella Strains Re-Emphasize
Zhong et al. Biotechnol Biofuels (2018) 11:193 https://doi.org/10.1186/s13068-018-1201-1 Biotechnology for Biofuels RESEARCH Open Access Pan‑genome analyses of 24 Shewanella strains re‑emphasize the diversifcation of their functions yet evolutionary dynamics of metal‑reducing pathway Chaofang Zhong, Maozhen Han, Shaojun Yu, Pengshuo Yang, Hongjun Li and Kang Ning* Abstract Background: Shewanella strains are important dissimilatory metal-reducing bacteria which are widely distributed in diverse habitats. Despite eforts to genomically characterize Shewanella, knowledge of the molecular components, functional information and evolutionary patterns remain lacking, especially for their compatibility in the metal- reducing pathway. The increasing number of genome sequences of Shewanella strains ofers a basis for pan-genome studies. Results: A comparative pan-genome analysis was conducted to study genomic diversity and evolutionary relation- ships among 24 Shewanella strains. Results revealed an open pan-genome of 13,406 non-redundant genes and a core-genome of 1878 non-redundant genes. Selective pressure acted on the invariant members of core genome, in which purifying selection drove evolution in the housekeeping mechanisms. Shewanella strains exhibited extensive genome variability, with high levels of gene gain and loss during the evolution, which afected variable gene sets and facilitated the rapid evolution. Additionally, genes related to metal reduction were diversely distributed in Shewanella strains and evolved under purifying selection, which highlighted the basic conserved functionality and specifcity of respiratory systems. Conclusions: The diversity of genes present in the accessory and specifc genomes of Shewanella strains indicates that each strain uses diferent strategies to adapt to diverse environments. -

Comprehensive Comparative Genomics Reveals Over 50 Phyla of Free‑Living and Pathogenic Bacteria Are Associated with Diverse Members of the Amoebozoa Yonas I
www.nature.com/scientificreports OPEN Comprehensive comparative genomics reveals over 50 phyla of free‑living and pathogenic bacteria are associated with diverse members of the amoebozoa Yonas I. Tekle*, Janae M. Lyttle, Maya G. Blasingame & Fang Wang The Amoebozoa, a group containing predominantly amoeboid unicellular protists has been shown to play an important ecological role in controlling environmental bacteria. Amoebozoans not only graze bacteria but also serve as a safe niche for bacterial replication and harbor endosymbiotic bacteria including dangerous human pathogens. Despite their importance, only a few lineages of Amoebozoa have been studied in this regard. In this research, we conducted a comprehensive genomic and transcriptomic study with expansive taxon sampling by including representatives from the three known clades of the Amoebozoa. We used culture independent whole culture and single cell genomics/transcriptomics to investigate the association of bacteria with diverse amoebozoans. Relative to current published evidence, we recovered the largest number of bacterial phyla (64) and human pathogen genera (51) associated with the Amoebozoa. Using single cell genomics/ transcriptomics we were able to determine up to 24 potential endosymbiotic bacterial phyla, some potentially endosymbionts. This includes the majority of multi‑drug resistant pathogens designated as major public health threats. Our study demonstrates amoebozoans are associated with many more phylogenetically diverse bacterial phyla than previously recognized. It also shows that all amoebozoans are capable of harboring far more dangerous human pathogens than presently documented, making them of primal public health concern. Te study of microbial interactions is a complex and fascinating feld of research 1–3. Microorganisms occupy diverse ecological niches and are usually found in large communities that result in inherent interactions. -

Phylogeny Reveals Novel Hipa-Homologous Kinase Families
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.14.439929; this version posted April 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Phylogeny Reveals Novel HipA-Homologous Kinase Families and Toxin – Antitoxin Gene Organizations Kenn Gerdes1*, Rene Bærentsen2 & Ditlev E. Brodersen2* 1) Centre of Excellence for Bacterial Stress Response and Persistence, Section for Functional Genomics, Department of Biology, University of Copenhagen, DK-2200, Copenhagen N, Denmark. 2) Department of Molecular Biology and Genetics, Aarhus University, DK-8000 Aarhus C, Denmark. * To whom correspondence should be addressed: [email protected] or [email protected] Running Head: Phylogeny of High Persister A-Homologous Kinases Keywords: High Persister A, HipB, HipS, HipT, HIRAN, Stl, GltX, TrpS bioRxiv preprint doi: https://doi.org/10.1101/2021.04.14.439929; this version posted April 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Toxin – Antitoxin modules function in the genetic stability of mobile genetic elements, bacteriophage defense, and antibiotic tolerance. A gain-of-function mutation of the Escherichia coli K-12 hipBA module can induce antibiotic tolerance in a subpopulation of bacterial cells, a phenomenon known as persistence. HipA is a Ser/Thr kinase that phosphorylates and inactivates glutamyl tRNA synthetase, inhibiting cellular translation and inducing the stringent response. -

Electron Donors and Acceptors for Members of the Family Beggiatoaceae
Electron donors and acceptors for members of the family Beggiatoaceae Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften - Dr. rer. nat. - dem Fachbereich Biologie/Chemie der Universit¨at Bremen vorgelegt von Anne-Christin Kreutzmann aus Hildesheim Bremen, November 2013 Die vorliegende Doktorarbeit wurde in der Zeit von Februar 2009 bis November 2013 am Max-Planck-Institut f¨ur marine Mikrobiologie in Bremen angefertigt. 1. Gutachterin: Prof. Dr. Heide N. Schulz-Vogt 2. Gutachter: Prof. Dr. Ulrich Fischer 3. Pr¨uferin: Prof. Dr. Nicole Dubilier 4. Pr¨ufer: Dr. Timothy G. Ferdelman Tag des Promotionskolloquiums: 16.12.2013 To Finn Summary The family Beggiatoaceae comprises large, colorless sulfur bacteria, which are best known for their chemolithotrophic metabolism, in particular the oxidation of re- duced sulfur compounds with oxygen or nitrate. This thesis contributes to a more comprehensive understanding of the physiology and ecology of these organisms with several studies on different aspects of their dissimilatory metabolism. Even though the importance of inorganic sulfur substrates as electron donors for the Beggiatoaceae has long been recognized, it was not possible to derive a general model of sulfur compound oxidation in this family, owing to the fact that most of its members can currently not be cultured. Such a model has now been developed by integrating information from six Beggiatoaceae draft genomes with available literature data (Section 2). This model proposes common metabolic pathways of sulfur compound oxidation and evaluates whether the involved enzymes are likely to be of ancestral origin for the family. In Section 3 the sulfur metabolism of the Beggiatoaceae is explored from a dif- ferent perspective. -

Metagenome-Assembled Genomes Provide New Insight Into The
www.nature.com/scientificreports Corrected: Author Correction OPEN Metagenome-assembled genomes provide new insight into the microbial diversity of two thermal Received: 17 August 2018 Accepted: 17 January 2019 pools in Kamchatka, Russia Published online: 28 February 2019 Laetitia G. E. Wilkins1,2, Cassandra L. Ettinger 2, Guillaume Jospin2 & Jonathan A. Eisen 2,3,4 Culture-independent methods have contributed substantially to our understanding of global microbial diversity. Recently developed algorithms to construct whole genomes from environmental samples have further refned, corrected and revolutionized understanding of the tree of life. Here, we assembled draft metagenome-assembled genomes (MAGs) from environmental DNA extracted from two hot springs within an active volcanic ecosystem on the Kamchatka peninsula, Russia. This hydrothermal system has been intensively studied previously with regard to geochemistry, chemoautotrophy, microbial isolation, and microbial diversity. We assembled genomes of bacteria and archaea using DNA that had previously been characterized via 16S rRNA gene clone libraries. We recovered 36 MAGs, 29 of medium to high quality, and inferred their placement in a phylogenetic tree consisting of 3,240 publicly available microbial genomes. We highlight MAGs that were taxonomically assigned to groups previously underrepresented in available genome data. This includes several archaea (Korarchaeota, Bathyarchaeota and Aciduliprofundum) and one potentially new species within the bacterial genus Sulfurihydrogenibium. Putative functions in both pools were compared and are discussed in the context of their diverging geochemistry. This study adds comprehensive information about phylogenetic diversity and functional potential within two hot springs in the caldera of Kamchatka. Terrestrial hydrothermal systems are of great interest to the general public and to scientists alike due to their unique and extreme conditions. -

Supplement of Biogeosciences, 16, 4229–4241, 2019 © Author(S) 2019
Supplement of Biogeosciences, 16, 4229–4241, 2019 https://doi.org/10.5194/bg-16-4229-2019-supplement © Author(s) 2019. This work is distributed under the Creative Commons Attribution 4.0 License. Supplement of Identifying the core bacterial microbiome of hydrocarbon degradation and a shift of dominant methanogenesis pathways in the oil and aqueous phases of petroleum reservoirs of different temperatures from China Zhichao Zhou et al. Correspondence to: Ji-Dong Gu ([email protected]) and Bo-Zhong Mu ([email protected]) The copyright of individual parts of the supplement might differ from the CC BY 4.0 License. 1 Supplementary Data 1.1 Characterization of geographic properties of sampling reservoirs Petroleum fluids samples were collected from eight sampling sites across China covering oilfields of different geological properties. The reservoir and crude oil properties together with the aqueous phase chemical concentration characteristics were listed in Table 1. P1 represents the sample collected from Zhan3-26 well located in Shengli Oilfield. Zhan3 block region in Shengli Oilfield is located in the coastal area from the Yellow River Estuary to the Bohai Sea. It is a medium-high temperature reservoir of fluvial face, made of a thin layer of crossed sand-mudstones, pebbled sandstones and fine sandstones. P2 represents the sample collected from Ba-51 well, which is located in Bayindulan reservoir layer of Erlian Basin, east Inner Mongolia Autonomous Region. It is a reservoir with highly heterogeneous layers, high crude oil viscosity and low formation fluid temperature. It was dedicated to water-flooding, however, due to low permeability and high viscosity of crude oil, displacement efficiency of water-flooding driving process was slowed down along the increase of water-cut rate.