HBO1-MLL Interaction Promotes AF4/ ENL/P-Tefb-Mediated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rabbit Anti-TAF1C/FITC Conjugated Antibody
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-TAF1C/FITC Conjugated antibody SL24053R-FITC Product Name: Anti-TAF1C/FITC Chinese Name: FITC标记的TATA盒Binding protein相关因子TAF1C抗体 RNA polymerase I-specific TBP-associated factor 110 kDa; SL1; Taf1c; TAF1C_MOUSE; TAFI110; TAFI95; TATA box binding protein associated factor 1C; TATA box-binding protein-associated factor 1C; TATA box-binding protein-associated Alias: factor RNA polymerase I subunit C; TBP associated factor 1C; TBP associated factor RNA polymerase I 95 kDa; TBP associated factor, RNA polymerase I, 110-KD; TBP- associated factor 1C; Transcription initiation factor SL1/TIF-IB subunit C. Organism Species: Rabbit Clonality: Polyclonal React Species: ICC=1:50-200IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 95kDa Form: Lyophilized or Liquid Concentration: 2mg/1ml immunogen: KLHwww.sunlongbiotech.com conjugated synthetic peptide derived from mouse TAF1C Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: Initiation of transcription by RNA polymerase I requires the formation of a complex Product Detail: composed of the TATA-binding protein (TBP) and three TBP-associated factors (TAFs) specific for RNA polymerase I. -

Cross-Cohort Analysis Identifies a TEAD4– MYCN Positive Feedback Loop As the Core Regulatory Element of High-Risk Neuroblastoma
Cross-Cohort Analysis Identifies a TEAD4– MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Rajbhandari, Presha et al. “Cross-Cohort Analysis Identifies a TEAD4–MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma.” Cancer Discovery 8, 5 (March 2018): 582–599 © 2018 AACR As Published http://dx.doi.org/10.1158/2159-8290.CD-16-0861 Publisher American Association for Cancer Research Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/117503 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Cancer Manuscript Author Discov. Author manuscript; Manuscript Author available in PMC 2018 August 01. Published in final edited form as: Cancer Discov. 2018 May ; 8(5): 582–599. doi:10.1158/2159-8290.CD-16-0861. Cross-cohort analysis identifies a TEAD4 ↔ MYCN positive- feedback loop as the core regulatory element of high-risk neuroblastoma Presha Rajbhandari1,2,$, Gonzalo Lopez1,3,$, Claudia Capdevila1, Beatrice Salvatori1, Jiyang Yu1,#, Ruth Rodriguez-Barrueco4,5, Daniel Martinez3, Mark Yarmarkovich3, Nina Weichert-Leahey6, Brian J. Abraham7, Mariano J Alvarez1, Archana Iyer1, Jo Lynne Harenza3, Derek Oldridge3, Katleen De Preter8, Jan Koster9, Shahab Asgharzadeh10,11, Robert -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

CASC3 Promotes Transcriptome-Wide Activation of Nonsense
bioRxiv preprint doi: https://doi.org/10.1101/811018; this version posted October 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 2 CASC3 promotes transcriptome-wide activation of nonsense- 3 mediated decay by the exon junction complex 4 5 Jennifer V. Gerbracht1, Volker Boehm1, Thiago Britto-Borges2,3, Sebastian Kallabis4, Janica L. 6 Wiederstein4, Simona Ciriello1,5, Dominik U. Aschemeier1, Marcus Krüger4, Christian K. 7 Frese4,6, Janine Altmüller7,8, Christoph Dieterich2,3, Niels H. Gehring1 8 1 Institute for Genetics, University of Cologne, 50674 Cologne, Germany 9 2 Section of Bioinformatics and Systems Cardiology, Department of Internal Medicine III and Klaus 10 Tschira Institute for Integrative Computational Cardiology, University of Heidelberg, 69120 Heidelberg, 11 Germany 12 3 DZHK (German Centre for Cardiovascular Research), Partner site Heidelberg/Mannheim, 69120 13 Heidelberg, Germany 14 4 CECAD Research Center, University of Cologne, Joseph-Stelzmann-Str. 26, 50931 Cologne, Germany 15 5 present address: AO Research Institute Davos, Clavadelerstrasse 8, CH-7270 Davos Platz, Switzerland 16 6 present address: Max Planck Unit for the Science of Pathogens, 10117 Berlin, Germany 17 7 Cologne Center for Genomics (CCG), University of Cologne, 50931 Cologne, Germany 18 8 Center for Molecular Medicine Cologne, University of Cologne, 50937 Cologne, Germany 19 20 21 Contact 22 Niels H. Gehring, University of Cologne, Institute for Genetics, Zuelpicher Str. 47a, 50674 Cologne, 23 Germany; email: [email protected] 24 25 26 27 Running Title (40 Characters) 28 CASC3 promotes EJC-dependent NMD 29 Keywords (5, alphabetical order, separated by slash) 30 CRISPR-Cas9/gene expression/mRNA quality control/NMD/RNA degradation 31 1 bioRxiv preprint doi: https://doi.org/10.1101/811018; this version posted October 21, 2019. -

Full-Text.Pdf
Systematic Evaluation of Genes and Genetic Variants Associated with Type 1 Diabetes Susceptibility This information is current as Ramesh Ram, Munish Mehta, Quang T. Nguyen, Irma of September 23, 2021. Larma, Bernhard O. Boehm, Flemming Pociot, Patrick Concannon and Grant Morahan J Immunol 2016; 196:3043-3053; Prepublished online 24 February 2016; doi: 10.4049/jimmunol.1502056 Downloaded from http://www.jimmunol.org/content/196/7/3043 Supplementary http://www.jimmunol.org/content/suppl/2016/02/19/jimmunol.150205 Material 6.DCSupplemental http://www.jimmunol.org/ References This article cites 44 articles, 5 of which you can access for free at: http://www.jimmunol.org/content/196/7/3043.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 23, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Systematic Evaluation of Genes and Genetic Variants Associated with Type 1 Diabetes Susceptibility Ramesh Ram,*,† Munish Mehta,*,† Quang T. -

Granulocyte Colony-Stimulating Factor-Induced TAF9 Modulates P53-TRIAP1-CASP3 Axis to Prevent Retinal Ganglion Cell Death After Optic Nerve Ischemia
Granulocyte Colony-Stimulating Factor-Induced TAF9 Modulates P53-TRIAP1-CASP3 Axis to Prevent Retinal Ganglion Cell Death after Optic Nerve Ischemia Yao-Tseng Wen Buddhist Tzu Chi General Hospital Hualien: Hualien Tzu Chi Hospital Keh-Liang Lin Chung Shan Medical University Hospital Chin-Te Huang Tzu Chi University Rong Kung Tsai ( [email protected] ) Hualien Tzu Chi Hospital https://orcid.org/0000-0001-8760-5739 Research article Keywords: Granulocyte colony-stimulating factor, Rat anterior ischemic optic neuropathy model, Retinal ganglion cell, TBP associated factor 9, TP53, TRIAP1 Posted Date: January 6th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-138908/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/25 Abstract Background Optic nerve head (ONH) infarct can result in progressive retinal ganglion cell (RGC) death. Some evidences indicated that the granulocyte colony-stimulating factor (GCSF) provides positive effects against ischemic damage on RGCs. However, protective mechanisms of the GCSF after ONH infarct are complex and remain unclear. Methods To investigate the complex mechanisms, the transcriptome proles of the GCSF-treated retinas were examined using microarray technology. The retinal mRNA samples on days 3 and 7 post rat anterior ischemic optic neuropathy model (rAION) were analyzed by microarray and bioinformatics analyses. To evaluate the TAF9 function in RGC apoptosis, GCSF plus TAF9 siRNA-treated rats were evaluated using retrograde labeling with FluoroGold assay, TUNEL assay, and Western blotting in a rAION. Results GCSF treatment inuenced 3101 genes and 3332 genes on days 3 and 7 post rAION, respectively. -

Setd1 Histone 3 Lysine 4 Methyltransferase Complex Components in Epigenetic Regulation
SETD1 HISTONE 3 LYSINE 4 METHYLTRANSFERASE COMPLEX COMPONENTS IN EPIGENETIC REGULATION Patricia A. Pick-Franke Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Master of Science in the Department of Biochemistry and Molecular Biology Indiana University December 2010 Accepted by the Faculty of Indiana University, in partial fulfillment of the requirements for the degree of Master of Science. _____________________________________ David Skalnik, Ph.D., Chair _____________________________________ Kristin Chun, Ph.D. Master’s Thesis Committee _____________________________________ Simon Rhodes, Ph.D. ii DEDICATION This thesis is dedicated to my sons, Zachary and Zephaniah who give me great joy, hope and continuous inspiration. I can only hope that I successfully set a good example demonstrating that one can truly accomplish anything, if you never give up and reach for your dreams. iii ACKNOWLEDGEMENTS I would like to thank my committee members Dr. Skalnik, Dr. Chun and Dr. Rhodes for allowing me to complete this dissertation. They have been incredibly generous with their flexibility. I must make a special thank you to Jeanette McClintock, who willingly gave her expertise in statistical analysis with the Cfp1 microarray data along with encouragement, support and guidance to complete this work. I would like to thank Courtney Tate for her ceaseless willingness to share ideas, and her methods and materials, and Erika Dolbrota for her generous instruction as well as the name of a good doctor. I would also like to acknowledge the superb mentorship of Dr. Jeon Heong Lee, PhD and the contagious passion and excitement for the life of science of Dr. -

Structural Mechanism of ATP-Independent Transcription
RESEARCH ARTICLE Structural mechanism of ATP-independent transcription initiation by RNA polymerase I Yan Han1, Chunli Yan2,3‡, Thi Hoang Duong Nguyen4‡, Ashleigh J Jackobel5, Ivaylo Ivanov2,3, Bruce A Knutson5*, Yuan He1* 1Department of Molecular Biosciences, Northwestern University, Evanston, United States; 2Department of Chemistry, Georgia State University, Atlanta, United States; 3Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, United States; 4Howard Hughes Medical Institute, University of California, Berkeley, United States; 5Department of Biochemistry and Molecular Biology, SUNY Upstate Medical University, Syracuse, United States Abstract Transcription initiation by RNA Polymerase I (Pol I) depends on the Core Factor (CF) complex to recognize the upstream promoter and assemble into a Pre-Initiation Complex (PIC). Here, we solve a structure of Saccharomyces cerevisiae Pol I-CF-DNA to 3.8 A˚ resolution using single-particle cryo-electron microscopy. The structure reveals a bipartite architecture of Core Factor and its recognition of the promoter from À27 to À16. Core Factor’s intrinsic mobility correlates well with different conformational states of the Pol I cleft, in addition to the stabilization of either Rrn7 N-terminal domain near Pol I wall or the tandem winged helix domain of A49 at a partially overlapping location. Comparison of the three states in this study with the Pol II system *For correspondence: knutsonb@ suggests that a ratchet motion of the Core Factor-DNA sub-complex at upstream facilitates upstate.edu (BAK); yuanhe@ promoter melting in an ATP-independent manner, distinct from a DNA translocase actively northwestern.edu (YHe) threading the downstream DNA in the Pol II PIC. -

Interstitial Deletion at 11Q14.2-11Q22.1 May Cause
Papoulidis et al. Molecular Cytogenetics (2015) 8:71 DOI 10.1186/s13039-015-0175-y CASE REPORT Open Access Interstitial deletion at 11q14.2-11q22.1 may cause severe learning difficulties, mental retardation and mild heart defects in 13-year old male Ioannis Papoulidis1*, Vassilis Paspaliaris1, Elisavet Siomou1, Sandro Orru2, Roberta Murru2, Stavros Sifakis3, Petros Nikolaidis4, Antonios Garas5, Sotirios Sotiriou5, Loretta Thomaidis6 and Emmanouil Manolakos1,2 Abstract Interstitial deletions of the long arm of chromosome 11 are rare, and they could be assumed as non-recurrent chromosomal rearrangements due to high variability of the size and the breakpoints of the deleted region. The exact region of the deletion was difficult to be determined before the use of molecular cytogenetic techniques such as array comparative genomic hybridization (aCGH). Here, a 13-year old boy with severe learning difficulties, mental retardation and mild heart defects is described. Conventional G-band karyotyping was performed and it is found that the patient is a carrier of a de novo interstitial deletion on the long arm of chromosome 11, involving 11q14 and 11q22 breakpoints. Further investigation, using aCGH, specified the deleted region to 11q14.2-11q22.1. There was a difficulty in correlating the genotype with the phenotype of the patient due to lack of similar cases in literature. More studies should be done in order to understand the genetic background that underlies the phenotypic differences observed in similar cases. Background Moreover, before the introduction of molecular cyto- Terminal deletions of the long arm of chromosome genetic approaches, the resolution efficiency provided 11 have been numerously described, and they are as- by conventional karyotype analysis jointly with the sym- sociated with Jacobsen syndrome (OMIM 147791) metric 11q banding pattern [3, 4], limited the accuracy and characterized by thrombocytopenia, mental re- of identification of breakpoints and precise deleted gen- tardation, short stature, congenital heart defect, and omic regions. -
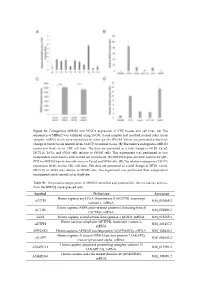
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

Network-Based Analysis of Key Regulatory Genes Implicated in Type
www.nature.com/scientificreports OPEN Network‑based analysis of key regulatory genes implicated in Type 2 Diabetes Mellitus and Recurrent Miscarriages in Turner Syndrome Anam Farooqui1, Alaa Alhazmi2, Shaful Haque3, Naaila Tamkeen4, Mahboubeh Mehmankhah1, Safa Tazyeen1, Sher Ali5 & Romana Ishrat1* The information on the genotype–phenotype relationship in Turner Syndrome (TS) is inadequate because very few specifc candidate genes are linked to its clinical features. We used the microarray data of TS to identify the key regulatory genes implicated with TS through a network approach. The causative factors of two common co‑morbidities, Type 2 Diabetes Mellitus (T2DM) and Recurrent Miscarriages (RM), in the Turner population, are expected to be diferent from that of the general population. Through microarray analysis, we identifed nine signature genes of T2DM and three signature genes of RM in TS. The power‑law distribution analysis showed that the TS network carries scale‑free hierarchical fractal attributes. Through local‑community‑paradigm (LCP) estimation we fnd that a strong LCP is also maintained which means that networks are dynamic and heterogeneous. We identifed nine key regulators which serve as the backbone of the TS network. Furthermore, we recognized eight interologs functional in seven diferent organisms from lower to higher levels. Overall, these results ofer few key regulators and essential genes that we envisage have potential as therapeutic targets for the TS in the future and the animal models studied here may prove useful in the validation of such targets. Te medical systems and scientists throughout the world are under an unprecedented challenge to meet the medical needs of much of the world’s population that are sufering from chromosomal anomalies. -

Bioinformatic Gene Analysis for Potential Therapeutic Targets Of
Xiang et al. J Transl Med (2020) 18:388 https://doi.org/10.1186/s12967-020-02549-9 Journal of Translational Medicine RESEARCH Open Access Bioinformatic gene analysis for potential therapeutic targets of Huntington’s disease in pre-symptomatic and symptomatic stage Chunchen Xiang1†, Shengri Cong1†, Bin Liang2 and Shuyan Cong1* Abstract Background: Huntington’s disease (HD) is a neurodegenerative disorder characterized by psychiatric symptoms, serious motor and cognitive defcits. Certain pathological changes can already be observed in pre-symptomatic HD (pre-HD) patients; however, the underlying molecular pathogenesis is still uncertain and no efective treatments are available until now. Here, we reanalyzed HD-related diferentially expressed genes from the GEO database between symptomatic HD patients, pre-HD individuals, and healthy controls using bioinformatics analysis, hoping to get more insight in the pathogenesis of both pre-HD and HD, and shed a light in the potential therapeutic targets of the disease. Methods: Pre-HD and symptomatic HD diferentially expressed genes (DEGs) were screened by bioinformatics analy- sis Gene Expression Omnibus (GEO) dataset GSE1751. A protein–protein interaction (PPI) network was used to select hub genes. Subsequently, Gene Ontology (GO) enrichment analysis of DEGs and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of hub genes were applied. Dataset GSE24250 was downloaded to verify our hub genes by the Kaplan–Meier method using Graphpad Prism 5.0. Finally, target miRNAs of intersected hub genes involved in pre- HD and symptomatic HD were predicted. Results: A total of 37 and 985 DEGs were identifed in pre-HD and symptomatic HD, respectively. The hub genes, SIRT1, SUZ12, and PSMC6, may be implicated in pre-HD, and the hub genes, FIS1, SIRT1, CCNH, SUZ12, and 10 others, may be implicated in symptomatic HD.